
Genetic Testing for FMR1 Mutations (including Fragile X Syndrome) - CAM 348
Description/Background
Fragile X syndrome (FXS) is an X-linked disorder resulting from a loss of function mutation of the Fragile X Mental Retardation-1 (FMR1) gene;1 FXS is the most common cause of heritable intellectual disability.2 FMR1-related disorders include FXS, fragile X-associated tremor/ataxia syndrome (FXTAS), and FMR1-related primary ovarian insufficiency (FRPOI). FXS results in a range of physical, cognitive, and behavioral effects of variable severity,3 generally characterized by moderate intellectual disability and autistic characteristics in affected males and mild intellectual disability and emotional and/or psychiatric problems in affected females.3,4
Terms such as male and female are used when necessary to refer to sex assigned at birth. For guidance on prenatal or pre-conception screening for FXS, please see CAM 358-Prenatal Screening (Genetic).
Regulatory Status
A search of the FDA database on Sept. 23, 2020, using the term “FMR1” yielded 0 results. Many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
- For individuals who have received genetic counseling, diagnostic genetic testing for FMR1 gene CGG repeats (including AGG interruption testing) and methylation status is considered MEDICALLY NECESSARY for any of the following conditions:
- For individuals with unexplained mental retardation, developmental delay, or autism spectrum disorder
- For symptomatic individuals with features of Fragile X syndrome or a family history of Fragile X syndrome.
- For females with unexplained ovarian insufficiency, unexplained ovarian failure, or unexplained elevated FSH prior to 40 years of age.
- For individuals with unexplained late-onset tremor-ataxia.
- For fetuses and offspring of known FMR1 premutation or full mutation carriers.
- Genetic screening for FMR1 gene CGG repeat length more than once per lifetime is considered NOT MEDICALLY NECESSARY.
- Determination of FMR1 gene point mutations is considered NOT MEDICALLY NECESSARY.
- Determination of FMR1 gene deletion is considered NOT MEDICALLY NECESSARY.
- General population screening for Fragile X syndrome is considered NOT MEDICALLY NECESSARY.
- Cytogenetic testing for Fragile X syndrome is considered NOT MEDICALLY NECESSARY..
- Testing for the FMRP protein is considered NOT MEDICALLY NECESSARY.
Table of Terminology
| Term |
Definition |
| ACMG |
American College of Medical Genetics and Genomics |
| ACOG |
The American College of Obstetricians and Gynecologists |
| ASD |
Autism Spectrum Disorder |
| CCMG |
Canadian College of Medical Geneticists |
| CI |
Confidence interval |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CMS |
Centers for Medicare and Medicaid |
| CNS |
Central nervous system |
| DBS |
Dried blood spots |
| dTP-PCR |
Triplet-primed polymerase chain reaction |
| EMQN |
European Molecular Genetics Quality Network |
| FDA |
Food and Drug Administration |
| FMR1 |
Fragile X mental retardation-1 |
| FMRP |
Protein encoded by Fragile X mental retardation-1 |
| FRPOI |
Fragile X mental retardation-1-Related Primary Ovarian Insufficiency |
| FSH |
Follicle stimulating hormone |
| FXS |
Fragile X syndrome |
| FXTAS |
Fragile X-associated tremor/ataxia syndrome |
| GDD |
Global Developmental Delay |
| ID |
Intellectual Disability |
| LDT |
Laboratory-developed test |
| mGluR |
Metabotropic glutamate receptor 2 |
| mRNA |
Messenger ribonucleic acid |
| miRNA |
Micro ribonucleic acid |
| MoMe |
Methylation mosaic |
| MoMN |
Mosaicism for full mutation and normal alleles |
| MoMP |
Mosaic premutation and full mutation |
| mRNA |
Messenger ribonucleic acid |
| NORD |
National Organization for Rare Disorders\ |
| PCR |
Polymerase chain reaction |
| RNA |
Ribonucleic acid |
| SB |
Southern blotting |
| SNP |
Single nucleotide polymorphisms |
| SOGC |
Society of Obstetricians and Gynecologists of Canada |
Rationale
Fragile X Syndrome (FXS) and related disorders affects about one in 4,000 males and one in 6,000 to 8,000 females in America.5 Transmitted as an X-linked dominant trait with reduced penetrance, FXS is associated with a fragile site on the X chromosome identified as the Fragile X Mental Retardation-1 (FMR1) gene.6,7 More than 99% of patients with FXS have a mutation in this gene with over 200 CGG repeats and atypical methylation.5 The protein encoded by FMR1 (FMRP) is a multifunctional RNA-binding protein that regulates the translation of a subset of dendritic mRNAs and plays a central role in neuronal development and synaptic plasticity.8-15 The absence of FMRP results in excessive and persistent mGluR-mediated protein synthesis in postsynaptic dendrites, dysregulation of ion homeostasis, and disruption of calcium ion homeostasis leading to abnormal synaptic signaling and dendritic development.13,16,17 The typical clinical phenotype includes intellectual disability, social impairment, autism spectrum disorder, speech and language delay, neurological dysfunction (seizures and abnormal sleep patterns), sensory hypersensitivity,18 and a characteristic physical appearance that typically develops in the second decade of life.19 Autism disorders are seen in approximately one third of FXS patients, affecting males more frequently than females.20 Between 55 and 90% of patients with autism and FXS report gaze aversion, hand flapping, repetitive behaviors, reduced social interaction, anxiety, speech preservations, and aggressive behaviors.21
Any genetic alteration that results in a lack of functional FMRP can cause FXS symptoms. The most common type of mutation of FMR1 is the expansion of a CGG trinucleotide repeat in the 5′ untranslated region of the gene.22 Normally, this ranges in size from seven to about 60 repeats, with 30 being most common.23 The full mutation consists of expansions of over 200 repeats which become abnormally hypermethylated, silencing the FMR1 gene and expression of FMRP.24,25 Molecular clinical correlations have shown that the resulting phenotype is related to the degree of methylation and mosaicism rather than the number of repeats.19
Alleles with 55 to 200 CGG repeats are generally unmethylated with normal transcript and FMRP level; however, they are extremely unstable during transmission to the next generation and are referred to as premutations.26 Although premutation carriers produce normal levels of FMRP, mRNA levels are elevated, causing toxic effects such as protein sequestration and mitochondrial dysfunction.27,28 As a consequence, RNA toxicity leads to neuronal toxicity and a spectrum of premutation associated disorders such as primary ovarian insufficiency (FXPOI) and tremor-ataxia syndrome (FXTAS).26,29 An increased frequency of neurological, psychological, endocrine, and immune-related characteristics has been documented in premutation carriers.30,31 Those with the premutation have higher rates of anxiety, depression, autistic traits, and physical health symptoms such as chronic fatigue and pain, fibromyalgia, and sleep disorders.32
It has been found that AGG interruptions (when an error in DNA replication results in an AGG interrupting the CGG-repeat tract with FMR1) may affect the stability of the fragile X triplet repeat in a positive manner. The presence of an AGG interruption has been found to substantially impact the risk of a full mutation expansion from a given repeat length. There is an emerging role for AGG genotyping to “clarify the course of fragile X genetic diagnosis, counseling, and patient management.”33 Others have also noted that the risk of unstable transmissions should be based on the presence or absence of AGG interruptions, not on the classical cutoffs which define the risk categories of FMR1 alleles.34
Analytical Validity
While many fragile X testing methods have been developed, no single approach can characterize all aspects of FMR1 mutations and expansions, especially when mosaicism is taken into consideration.4 In a diagnostic setting, it is important to not only detect presence of the CGG expansion, but to also determine its size and methylation status.35 Molecular diagnostic testing of FMR1 currently relies on a combination of polymerase chain reaction (PCR) and Southern blot (the gold standard) for the CGG-repeat expansion and methylation analyses.36,37 Detection of rare point mutations and deletions requires sequence analysis.38,39 This has limited the ability to implement any type of population screening.40
CGG repeat-primed PCR designed to detect the full range of fragile X expanded alleles followed by analysis via capillary electrophoresis and melt curve techniques minimizes the need for Southern blot analysis.41-44 The FastFraX FMR1 test was evaluated in 198 archived clinical samples, yielding results of 100% sensitivity (95% CI, 91.03% to 100%) and 100% specificity (95% CI, 97.64% to 100%) in categorizing patient samples into the respective normal, intermediate, premutation, and full mutation genotypes.35
The triplet-primed PCR method (dTP-PCR) has been validated by comparison to Southern blot analysis for use in determining mutations in the FMR1 gene; clinical performance was confirmed with 40 samples resulting in 100% sensitivity and 90.48% specificity in the detection of CGG repeats greater than 30.45 This testing method may be utilized to screen a general population by quickly determining specific allelic changes in the FMR1 gene.45
Immunohistochemical detection of FMRP has been validated in lymphocytes and chorionic villi samples as an alternative prenatal diagnostic method for detection of full mutations in male fetuses; however, staining is more complex in female fetuses due to X-inactivation and is insufficient for diagnostic use.46,47 Clinical and analytical specificity and sensitivity of cytogenetic analysis for FXS are both insufficient.4
In a retrospective design, Ramos, et al. (2020) studied the performance of the commercial FragileEase PCR kit for FXS diagnosis and compared it to Southern blotting (SB), PCR, and AmplideX FMR1 PCR. Ninety DNA samples were analyzed using FragileEase from patients with a clinical suspicion of FXS or a family history and was compare with the results from the other methods. Overall, FragileEase PCR kit had high concordance with the results from PCR, SB, and AmplideX. FragileEase was able to detect normal, intermediate/gray zone, premutation, and full mutation alleles along with female homozygosity and mosaicism. The authors conclude that "FragileEase™ PCR, as well as other commercially available kits, efficiently detect FMR1 mutations and simplify the workflow in laboratories that performing FXS diagnoses.”48
Clinical Utility and Validity
As the clinical phenotype of FMR-related diseases can be subtle, its detection, especially in the prepubertal period, can be difficult. Although phenotypic symptoms are not obvious at birth, both animal and neuroimaging studies suggest that the effects of FXS begin in the prenatal period.40 Families report significant delays in diagnosis of FXS with 24% of families reporting that they had seen a healthcare provider more than ten times before testing. On average, caregivers or other individuals first report concern in regard to the child's development by 13 months; however, professional confirmation of a developmental delay did not occur until an average age of 21 months, and the FXS diagnosis did not occur until an average age of nearly 32 months. Meanwhile, many families had additional children with FXS before becoming aware of the reproductive risk.49 Establishing a diagnosis of FXS allows for an understanding of the disorder and education on appropriate management strategies. Psychopharmacologic intervention to modify behavioral problems, such as attentional deficits, impulse control, anxiety, and emotional lability in a child with FXS can be important in addition to speech therapy, occupational therapy, special educational services, and behavioral interventions.19 A recent pilot of allopregnanolone in six males with FXTAS showed significant improvement in GABA metabolism, oxidative stress, and some of the mitochondria-related outcomes.50
Huang, et al. (2019) utilized a GC-rich PCR method to detect FMR1 gene mutations in 30 pregnant woman who were known carriers of FMR1 mutations or who contained FMR1 gene deletion mosaicism; samples utilized chorionic villus, amniotic fluid, or umbilical blood samples. Southern blotting was used as a confirmatory measure. PCR results showed that 18 fetuses were normal, while others presented with full FMR1 gene mutations, premutations, and/or mosaicism. Even with successful results, the authors state that the use of a single detention method may not be sufficient in determining FMR1 genetic mutations.51
Lee, et al. (2020) utilized a customized PCR and software system to detect the FMR1 gene expansions from dried blood spots (DBS) and performed analytical validity studies to determine its accuracy, specificity, sensitivity, and precision to be used for newborn screening. The study investigated 963 newborn DBS, which were studied by DNA extraction, FMR1 PCR amplification, and capillary electrophoresis for automated CGG-repeat analysis. While previous FMR1 newborn screening assays were unsuitable for a routine laboratory setting, this fit-for-purpose FMR1 screening method provides a reliable method for newborn screening that is both cost effective and compatible with simple DBS elution methods already used in newborn screening laboratories. From the 963 DBS samples tested, 957 samples (99.4%) samples were classified as normal and six samples (0.6%) had premutation alleles with 55-76 CGG-repeat expansions. Five out of the six premutation samples had one normal allele in addition to the premutation allele, while one out of the six had only one allele. Accuracy testing results were 100% concordance with reference genotypes with no false positive or false negative test results found. CGG expansions were consistently within six CGG repeats for larger expansions up to 200, within three CGG repeats for expansions up to 137, and within a single repeat for CGG expansions less than 80. However, the authors wrote that “further studies are required to identify if early screening of Fragile X syndrome would lead to better outcomes for the children, families, and society.”52
Approximately twenty individuals have been reported with rare missense or nonsense mutations in the FMR1 gene; also reported were other coding disturbances of the same gene resulting in physical, cognitive, and behavioral features similarly seen in FXS.39 Studies of other FMR mutations that can affect the level and function of the protein include analysis of SNPs showing that 31.66 % of the FMR1 gene SNPs were disease-related and that 50% of SNPs from online databases had a pathogenic effect.53 Screening of 508 males with clinical signs of mental retardation and developmental delay, but without CGG and GCC repeat expansions in the FMR1 gene, revealed two missense mutations in the FMR1 gene that would have not been diagnosed with standard molecular testing for FXS.54
Cao, et al. (2021) studied the clinical utility of screening FMR1 gene mutations during early and middle pregnancy for those carrying high-risk CGG trinucleotide expansions. DNA samples from, 316 pregnant women at 12-21 gestational weeks were collected and analyzed for CGG repeats using fluorescence PCR and capillary electrophoresis. The carrier rate of CGG repeats was one in 178 for the intermediate type and one in 772 for the premutation types. The highest frequency allele of CGG was 29 repeats, which accounted for 49.29%, followed by 30 repeats (28.56%) and 36 repeats (8.83%). In one case of a premutation type of CGG expansion, the couple chose to terminate the pregnancy. The authors conclude that "pregnant women should be screened for FMR1 gene mutations during early and middle pregnancy, and those with high-risk CGG expansions should undergo prenatal diagnosis, genetic counseling and family study.”55 Ramos, et al. (2021) conducted a cross-sectional study on FMR1 gene mutations in 52 Brazilian women diagnosed with primary ovarian insufficiency. The authors extracted genomic DNA and used FragileEase PCR kits to analyze CGG trinucleotide repeat expansions in the FMR1 gene. In total, 3.8% of participants had FMR1 mutations. The authors further concluded that “the most frequent CGG-repeat sizes were 28 and 30.”56
Fisher, et al. (2021) studied the predisposition of carriers to a neurodegenerative disease called Fragile X-associated tremor/ataxia syndrome (FXTAS). FXTAS is caused by “expansions of the CGG repeats in the 5’ upstream region of the FMR1 gene from the normal range.”57 The authors noted that individuals in the premutation group showed CGG expansion sizes with a peak in the 80-99 repeat size range. The two groups in the study included 33 participants who were controls and 41 participants who were FMR1 premutation carriers. The authors were interested in the role of mitochondrial dysfunction and associated cellular-stress signaling in carriers versus healthy control subjects. Results confirmed “the elevation of AMPK and mitochondrial respiratory activities and reduction in reactive O2 species (ROS) levels in premutation cells and revealed for the first time that target of rapamycin complex I (TORC1) activities are reduced.”57 This study also confirmed findings of a previous study in which they reported significant elevation of mitochondrial respiratory functions in FMR1 premutation carriers.
Lindstrand, et al. (2022) conducted a study comparing diagnostic methods in patients with intellectual disability and neurodevelopmental disorders using genome sequencing or chromosomal microarray with or without FMR1 analysis. From the genomic sequencing tests, when using it first, the diagnostic yield was 35%, 26% for when genomic sequencing was second, and 11% for CMA with or without FMR1 analysis. They also identified that costs were higher with CMA/FMR1, and that the majority (91%) of those with a negative result from the CMA/FMR1 analysis remain undiagnosed of definitive intellectual disability or neurodevelopmental disorder. This demonstrated that genome testing may be superior to traditional CMA and FMR1 analysis as a first-line test for those with neurocognitive difficulties, thus rendering it a faster and more cost effective method that is worth further investigating.58
American College of Medical Genetics and Genomics (ACMG)
The ACMG recommends FXS molecular genetic testing for:
Fragile X syndrome:
- Individuals of either sex with mental retardation, developmental delay, or autism, especially if they have (a) any physical or behavioral characteristics of fragile X syndrome, (b) a family history of fragile X syndrome, or (c) male or female relatives with undiagnosed mental retardation.
- Individuals seeking reproductive counseling who have (a) a family history of fragile X syndrome or (b) a family history of undiagnosed mental retardation.
- Fetuses of known carrier mothers.
- Affected individuals or their relatives in the context of a positive cytogenetic fragile X test result who are seeking further counseling related to the risk of carrier status among themselves or their relatives. The cytogenetic test was used prior to the identification of the FMR1 gene and is significantly less accurate than the current DNA test. DNA testing on such individuals is warranted to accurately identify premutation carriers and to distinguish premutation from full mutation carrier women.
Ovarian dysfunction:
- Women who are experiencing reproductive or fertility problems associated with elevated follicle stimulating hormone (FSH) levels, especially if they have (a) a family history of premature ovarian failure, (b) a family history of fragile X syndrome, or (c) male or female relatives with undiagnosed mental retardation.
Tremor/ataxia syndrome:
- Men and women who are experiencing late onset intention tremor and cerebellar ataxia of unknown origin, especially if they have (a) a family history of movement disorders, (b) a family history of fragile X syndrome, or (c) male or female relatives with undiagnosed mental retardation.”59
The 2013 ACMG fragile X testing standards and guidelines, with the American Congress of Obstetricians and Gynecologists, published the following indications for fragile X diagnostic testing and carrier detection:
- “The identification of a full mutation in a male is considered diagnostic rather than predictive, inasmuch as penetrance of fragile X syndrome is virtually 100% in males and the age of onset is not variable.
- The identification of a full mutation in a female may be diagnostic, but [over] 50% of females with full mutations have intellectual disability. They may, however, have some manifestations of the disease such as avoidance personality, mood, or stereotypic disorders. Nonrandom X inactivation may explain the milder phenotype in females, although the extent of symptoms cannot be determined by X-inactivation patterns from diagnostic tests that determine the expansion and methylation in blood.
- The identification of a premutation in an asymptomatic male or female undergoing carrier testing (e.g., due to a family history of intellectual disability) is predictive because FXPOI and FXTAS are not fully penetrant and are dependent on both age and allele size.
- All positive results should state that genetic counseling is recommended and testing is available for at-risk family members.”4
In 2021, ACMG released an updated guideline for screening for autosomal recessive and X-linked conditions during pregnancy and preconception. Their practice resource aims to recommend “a consistent and equitable approach for offering carrier screening to all individuals during pregnancy and preconception” and replaces any earlier ACMG position statements on prenatal/preconception expanded carrier screening and provide the following recommendations:
- “Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.”
- “The phrase “expanded carrier screening” be replaced by “carrier screening.””
- “Adopting a more precise tiered system based on carrier frequency:
- Tier 4: <1/200 carrier frequency (includes Tier 3) genes/condition will vary by lab
- Tier 3: ≥ 1/200 carrier frequency (includes Tier 2) includes X-linked conditions
- Tier 2: ≥1/100 carrier frequency (includes Tier 1)
- Tier 1: CF [Cystic Fibrosis] + SMA [spinal muscular atrophy] + Risk Based Screening”
- “Tier 1 screening conveys the recommendations previously adopted by ACMG and ACOG” and “adopts an ethnic and population neutral approach when screening for cystic fibrosis and spinal muscular atrophy. Beyond these two conditions, additional carrier screening is determined after risk assessment, which incorporates personal medical and family history as well as laboratory and imaging information where appropriate.”
- “Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.” However, “data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥1/100 creates missed opportunities to identify couples at risk for serious conditions.”
- “We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥1/200 . . . Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use “carrier frequency” to mean in any ethnic group with reasonable representation in the United States.”
- “Tier 4 includes genes less common than those in Tier 3 and can identify additional at-risk couples. Tier 4 has no lower limit carrier screening frequency and can greatly extend the number of conditions screened . . . the clinical validity at this level of carrier screening may be less compelling, therefore we suggest reserving this level of screening for consanguineous pregnancies (second cousins or closer) and in couples where family or medical history suggests Tier 4 screening might be beneficial . . . Importantly, patients should understand that their chance of being a carrier for one or more conditions increases as the number of conditions screened is increased.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.”
- Tier 4 screening should be considered:
- When a pregnancy stems from a known or possible consanguineous relationship (second cousins or closer);
- When a family or personal medical history warrants.
- ACMG does NOT recommend:
- Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
- Routine offering of Tier 4 panels.
- “Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for … X-linked (Table 6) conditions.”
- “All XX patients should be offered screening for only those X-linked genes listed in Table 6 as part of Tier 3 screening.”
- “When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered.”60
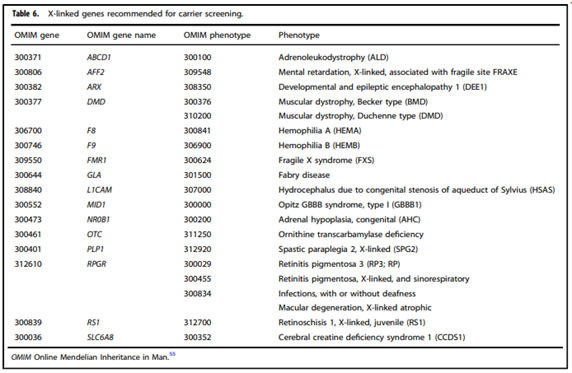
Table six from Gregg, et al. (2021)
The American College of Obstetricians and Gynecologists (ACOG)
The ACOG published committee opinion 691 in 2017, that was reaffirmed in 2023 which recommends fragile X premutation carrier screening for women with a family history of fragile X-related disorders or intellectual disability suggestive of FXS and who are considering pregnancy or are currently pregnant.61
If a woman has unexplained ovarian insufficiency or failure or an elevated FSH level before age 40 years, fragile X carrier screening is recommended to determine whether she has an FMR1 premutation.
All identified individuals with intermediate results and carriers of a fragile X premutation or full mutation should be provided follow-up genetic counseling to discuss the risk to their offspring of inheriting an expanded full mutation fragile X allele and to discuss fragile X-associated disorders (premature ovarian insufficiency and fragile X tremor/ataxia syndrome).
Prenatal diagnostic testing for FXS should be offered to known carriers of the fragile X premutation or full mutation.61
Society of Obstetricians and Gynecologists of Canada (SOGC) and Canadian College of Medical Geneticists (CCMG) Guidelines
Guidelines for FXS genetic testing were given in a joint statement from the SOGC and CCMG. It is stated that “any woman with a personal or family history of Fragile X- or Fragile X mental retardation 1–related disorders; unexplained intellectual disability or developmental delay; autism; ovarian insufficiency with elevated follicle stimulating hormone at age < 40 years of unknown etiology; or any woman with a family history of male relatives with developmental delay, autism, or isolated cerebellar ataxia and tremor should be offered screening for this condition (II-2A) (GRADE moderate/moderate).”62 It is also stated that “population carrier screening for Fragile X syndrome in all women of reproductive age cannot be recommended at this time (II-2D) (GRADE moderate/moderate)” and “Fragile X carrier testing must only occur after detailed genetic counselling and informed consent from the woman to be tested has been obtained (III-A) (GRADE low/moderate).”62 This statement has since been retired as of 2023. The most updated guidelines regarding prenatal screening for fetal aneuploidy, fetal anomalies, and adverse pregnancy outcomes as well as guidelines for using chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss from SOGC and CCMG do not make a direct mention of FXS genetic testing.63,64
Canadian College of Medical Geneticists (CCMG)
The CCMG released a position statement on Genetic and Metabolic Investigations for Neurodevelopmental Disorders, which includes updated recommendations for fragile X (FMR1) testing and outlines clinical features suggestive of FXS.
“Recommendations for fragile X testing
- FXS testing is recommended as a first-tier diagnostic test for individuals presenting with:
- GDD, ID or ASD and a clinical presentation or family history suggestive of FXS
- Any NDD and a family history of FXS or other FMR1-related disorder.
- FXS testing is not recommended for individuals with GDD or ID who do not meet the above criteria and have a complex clinical presentation that is not consistent with FXS (eg, multiple congenital anomalies, profound neurological impairment).”65
Clinical features that may be suggestive of fragile X syndrome
“Proband history and physical exam
- Macro-orchidism (may not be present until after puberty).
- Relative or mild (+2–3 SD) macrocephaly.
- Facial features: large or prominent ears, long or narrow face, tall forehead, high-arched palate and prominent jaw.
- Connective tissue findings: soft or velvety skin, redundant skin on dorsum of hands, hyperextensible joints, pes planus and mitral valve prolapse.
- Behaviour: ASD or autistic features, hyperactivity, shyness, gaze avoidance, hand biting, tactile defensiveness and anxiety.”65
“Family History in relatives on the maternal side:
- Males or females with GDD, ID or ASD.
- Females with premature menopause or ovarian insufficiency.
Males or females with adult-onset tremor, ataxia or parkinsonism.”65
National Society of Genetic Counselors (NSGC)
The NSGC published guidelines, which recommend: “Centers offering population screening should ensure that they have the resources available to provide pre- and post-test genetic counseling that supports the psychosocial and clinical needs of the patient and family. In light of widespread FMR1 testing among women without known risk factors, genetic counselors should anticipate seeing patients who did not receive any pre-test information, have no prior knowledge of FMR1-associated disorders, and are unprepared to learn that they have an FMR1 mutation. Prenatal diagnosis should be offered to women with pre- or full mutations. Males with premutation alleles should receive genetic counseling about potential phenotypic risks to their daughters, all of whom will inherit premutations.”17
American Academy of Pediatrics Committee on Genetics (AAP)
The American Academy of Pediatrics recommends testing for FXS in children with any of the following, particularly when associated with physical and behavioral characteristics of FXS or a relative with undiagnosed intellectual disability: developmental delay, borderline intellectual abilities or intellectual disability, or diagnosis of autism without a specific etiology.19
European Molecular Genetics Quality Network (EMQN)
The EMQN published their best practice guidelines concerning FXS and fragile X-associated disorders in 2015. They state, “Prenatal testing is not indicated for the pregnant partner of a male with a premutation.” but they do recommend offering prenatal diagnosis to any woman with 55 or more CGG repeats; “Prenatal testing may be considered for a female fetus of a full mutation father as a cautionary measure (full mutation or MoMP [mosaic premutation and full mutation] or MoMe [methylation mosaic]).” Concerning molecular diagnostic analysis in FXS and fragile X-associated disorders, they state the following:
“It is best practice to use a method which detects the whole range of expansions when testing relatives (including prenatal diagnosis) in a family with any known fragile X disorder due to expansion. When testing the FMR1 gene in population screening, the report must specify that rare cases of point mutation or deletion cannot be detected, nor rare cases of CGG expansion mosaicism (MoMN) if the method used cannot detect the whole range of expansions. It could be useful to confirm results by an independent method when detecting an expansion in an index case depending on specific pitfalls of each method.”66
References:
1. Saul RA, Tarleton JC. FMR1-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews((R)). 1993.
2. Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. Oct 2009;85(4):503-14. doi:10.1016/j.ajhg.2009.09.007
3. Mila M, Rodriguez-Revenga L, Matilla-Duenas A. FMR1 Premutation: Basic Mechanisms and Clinical Involvement. Cerebellum. Oct 2016;15(5):543-5. doi:10.1007/s12311-016-0808-7
4. Monaghan KG, Lyon E, Spector EB, American College of Medical GaG. ACMG Standards and Guidelines for fragile X testing: a revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet Med. Jul 2013;15(7):575-86. doi:10.1038/gim.2013.61
5. NORD. Fragile X Syndrome. Updated April 18, 2022. https://rarediseases.org/rare-diseases/fragile-x-syndrome/
6. Yu S, Pritchard M, Kremer E, et al. Fragile X genotype characterized by an unstable region of DNA. Science. May 24 1991;252(5009):1179-81. doi:10.1126/science.252.5009.1179
7. Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219-45. doi:10.1146/annurev-pathol-011811-132457
8. Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. May-Jun 2006;32(1-2):37-48. doi:10.1016/j.mcn.2006.02.001
9. Bechara EG, Didiot MC, Melko M, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. Jan 20 2009;7(1):e16. doi:10.1371/journal.pbio.1000016
10. Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL, Moine H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. Sep 2008;36(15):4902-12. doi:10.1093/nar/gkn472
11. Ascano M, Jr., Mukherjee N, Bandaru P, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. Dec 20 2012;492(7429):382-6. doi:10.1038/nature11737
12. Kenny PJ, Zhou H, Kim M, et al. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep. Dec 11 2014;9(5):1729-1741. doi:10.1016/j.celrep.2014.10.054
13. Castagnola S, Delhaye S, Folci A, et al. New Insights Into the Role of Cav2 Protein Family in Calcium Flux Deregulation in Fmr1-KO Neurons. Front Mol Neurosci. 2018;11:342. doi:10.3389/fnmol.2018.00342
14. Parvin S, Takeda R, Sugiura Y, Neyazaki M, Nogi T, Sasaki Y. Fragile X mental retardation protein regulates accumulation of the active zone protein Munc18-1 in presynapses via local translation in axons during synaptogenesis. Neurosci Res. Sep 19 2018;doi:10.1016/j.neures.2018.09.013
15. Yang YM, Arsenault J, Bah A, et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol Psychiatry. Sep 17 2018;doi:10.1038/s41380-018-0240-0
16. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. Jul 2004;27(7):370-7. doi:10.1016/j.tins.2004.04.009
17. Finucane B, Abrams L, Cronister A, Archibald AD, Bennett RL, McConkie-Rosell A. Genetic counseling and testing for FMR1 gene mutations: practice guidelines of the national society of genetic counselors. Journal of genetic counseling. Dec 2012;21(6):752-60. doi:10.1007/s10897-012-9524-8
18. Rais M, Binder DK, Razak KA, Ethell IM. Sensory Processing Phenotypes in Fragile X Syndrome. ASN Neuro. Jan-Dec 2018;10:1759091418801092. doi:10.1177/1759091418801092
19. Hersh JH, Saul RA. Health supervision for children with fragile X syndrome. Pediatrics. May 2011;127(5):994-1006. doi:10.1542/peds.2010-3500
20. Ormazabal M, Solari A, Espeche L, Castro T, Buzzalino N. [Fragile X syndrome and other entities associated with the FMR1 gene: Study of 28 affected families]. Arch Argent Pediatr. Jun 1 2019;117(3):e257-e262. Fragilidad del X y otras entidades asociadas al gen FMR1: estudio de 28 familias afectadas. doi:10.5546/aap.2019.e257
21. Reisinger DL, Shaffer RC, Tartaglia N, Berry-Kravis E, Erickson CA. Delineating Repetitive Behavior Profiles across the Lifespan in Fragile X Syndrome. Brain Sci. Apr 17 2020;10(4)doi:10.3390/brainsci10040239
22. Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. Apr 12 2000;9(6):901-8. doi:10.1093/hmg/9.6.901
23. Peprah E. Fragile X syndrome: the FMR1 CGG repeat distribution among world populations. Ann Hum Genet. Mar 2012;76(2):178-91. doi:10.1111/j.1469-1809.2011.00694.x
24. Maurin T, Zongaro S, Bardoni B. Fragile X Syndrome: from molecular pathology to therapy. Neurosci Biobehav Rev. Oct 2014;46 Pt 2:242-55. doi:10.1016/j.neubiorev.2014.01.006
25. Oberle I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. May 24 1991;252(5009):1097-102. doi:10.1126/science.252.5009.1097
26. Zafarullah M, Tang H-T, Durbin-Johnson B, et al. FMR1 locus isoforms: potential biomarker candidates in fragile X-associated tremor/ataxia syndrome (FXTAS). Scientific Reports. 2020/07/06 2020;10(1):11099. doi:10.1038/s41598-020-67946-y
27. Garcia-Arocena D, Hagerman PJ. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. Apr 15 2010;19(R1):R83-9. doi:10.1093/hmg/ddq166
28. Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. Jan 2000;66(1):6-15. doi:10.1086/302720
29. Rosario R, Anderson R. The molecular mechanisms that underlie fragile X-associated premature ovarian insufficiency: is it RNA or protein based? Molecular Human Reproduction. 2020;doi:10.1093/molehr/gaaa057
30. Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. Aug 2013;12(8):786-98. doi:10.1016/S1474-4422(13)70125-X
31. Raspa M, Wylie A, Wheeler AC, et al. Sensory Difficulties in Children With an FMR1 Premutation. Front Genet. 2018;9:351. doi:10.3389/fgene.2018.00351
32. Johnson K, Herring J, Richstein J. Fragile X Premutation Associated Conditions (FXPAC). Front Pediatr. 2020;8:266. doi:10.3389/fped.2020.00266
33. Latham GJ, Coppinger J, Hadd AG, Nolin SL. The role of AGG interruptions in fragile X repeat expansions: a twenty-year perspective. Front Genet. 2014;5:244. doi:10.3389/fgene.2014.00244
34. Villate O, Ibarluzea N, Maortua H, et al. Effect of AGG Interruptions on FMR1 Maternal Transmissions. Front Mol Biosci. 2020;7:135. doi:10.3389/fmolb.2020.00135
35. Lim GX, Yeo M, Koh YY, et al. Validation of a commercially available test that enables the quantification of the numbers of CGG trinucleotide repeat expansion in FMR1 gene. PLoS One. 2017;12(3):e0173279. doi:10.1371/journal.pone.0173279
36. Rajan-Babu IS, Chong SS. Molecular Correlates and Recent Advancements in the Diagnosis and Screening of FMR1-Related Disorders. Genes (Basel). Oct 14 2016;7(10)doi:10.3390/genes7100087
37. Cai X, Arif M, Wan H, Kornreich R, Edelmann LJ. Clinical Genetic Testing for Fragile X Syndrome by Polymerase Chain Reaction Amplification and Southern Blot Analyses. Methods Mol Biol. 2019;1942:11-27. doi:10.1007/978-1-4939-9080-1_2
38. Suhl JA, Warren ST. Single-Nucleotide Mutations in FMR1 Reveal Novel Functions and Regulatory Mechanisms of the Fragile X Syndrome Protein FMRP. J Exp Neurosci. 2015;9(Suppl 2):35-41. doi:10.4137/JEN.S25524
39. Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ, Butler MG. Rare FMR1 gene mutations causing fragile X syndrome: A review. Am J Med Genet A. Jan 2018;176(1):11-18. doi:10.1002/ajmg.a.38504
40. Riley C, Wheeler A. Assessing the Fragile X Syndrome Newborn Screening Landscape. Pediatrics. Jun 2017;139(Suppl 3):S207-S215. doi:10.1542/peds.2016-1159G
41. Lyon E, Laver T, Yu P, et al. A simple, high-throughput assay for Fragile X expanded alleles using triple repeat primed PCR and capillary electrophoresis. J Mol Diagn. Jul 2010;12(4):505-11. doi:10.2353/jmoldx.
42. Chen L, Hadd A, Sah S, et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn. Sep 2010;12(5):589-600. doi:10.2353/jmoldx.
43. Teo CR, Law HY, Lee CG, Chong SS. Screening for CGG repeat expansion in the FMR1 gene by melting curve analysis of combined 5' and 3' direct triplet-primed PCRs. Clin Chem. Mar 2012;58(3):568-79. doi:10.1373/clinchem.
44. Rajan-Babu IS, Teo CR, Lian M, Lee CG, Law HY, Chong SS. Single-tube methylation-specific duplex-PCR assay for rapid and accurate diagnosis of Fragile X Mental Retardation 1-related disorders. Expert Rev Mol Diagn. Mar 2015;15(3):431-41. doi:10.1586/14737159.2015.1001749
45. Skrlec I, Barisic K, Wagner J. Validation of a Screening Method for Dynamic Mutations in the FMR1 Gene. Ann Clin Lab Sci. Nov 2018;48(6):810-813.
46. Oostra BA, Willemsen R. Diagnostic tests for fragile X syndrome. Expert Rev Mol Diagn. Jul 2001;1(2):226-32. doi:10.1586/14737159.1.2.226
47. Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. Jun 2002;110(6):601-5. doi:10.1007/s00439-002-0723-5
48. Ramos C, Ocampos M, Barbato IT, Graça Bicalho Md, Nisihara R. Molecular analysis of FMR1 gene in a population in Southern Brazil: Comparison of four methods. Practical Laboratory Medicine. 2020/08/01/ 2020;21:e00162. doi:10.1016/j.plabm.2020.e00162
49. Bailey DB, Jr., Skinner D, Sparkman KL. Discovering fragile X syndrome: family experiences and perceptions. Pediatrics. Feb 2003;111(2):407-16. doi:10.1542/peds.111.2.407
50. Napoli E, Schneider A, Wang JY, et al. Allopregnanolone Treatment Improves Plasma Metabolomic Profile Associated with GABA Metabolism in Fragile X-Associated Tremor/Ataxia Syndrome: a Pilot Study. Mol Neurobiol. Sep 5 2018;doi:10.1007/s12035-018-1330-3
51. Huang W, Xue J, Kang H, et al. [Prenatal diagnosis for 30 women carrying a FMR1 mutation]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. Sep 10 2019;36(9):866-869.
52. Lee S, Taylor JL, Redmond C, et al. Validation of Fragile X Screening in the Newborn Population Using a Fit-for-Purpose FMR1 PCR Assay System. J Mol Diagn. Mar 2020;22(3):346-354. doi:10.1016/j.jmoldx.2019.11.002
53. Tekcan A. In Silico Analysis of FMR1 Gene Missense SNPs. Cell Biochem Biophys. Jun 2016;74(2):109-27. doi:10.1007/s12013-016-0722-0
54. Handt M, Epplen A, Hoffjan S, Mese K, Epplen JT, Dekomien G. Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol Cell Probes. Oct-Dec 2014;28(5-6):279-83. doi:10.1016/j.mcp.2014.08.003
55. Cao Q, Mu W, Sun D, et al. [Significance and case analysis of FMR1 mutation screening during early and middle pregnancy]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics.
56. Ramos C, Ocampos M, Barbato IT, Niehues VMS, Bicalho MDG, Nisihara R. Association between mutations in the FMR1 gene and ovarian dysfunction in Brazilian patients. JBRA Assist Reprod. Sep 20 2021;
57. Fisher PR, Allan CY, Sanislav O, et al. Relationships between Mitochondrial Function, AMPK, and TORC1 Signaling in Lymphoblasts with Premutation Alleles of the FMR1 Gene. Int J Mol Sci. Sep 27 2021;22(19)doi:10.3390/ijms221910393
58. Lindstrand A, Ek M, Kvarnung M, et al. Genome sequencing is a sensitive first-line test to diagnose individuals with intellectual disability. Genet Med. Nov 2022;24(11):2296-2307. doi:10.1016/j.gim.2022.07.022
59. Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. Oct 2005;7(8):584-7. doi:10.1097%2F01.GIM.0000182468.22666.dd
60. Gregg AR, Aarabi M, Klugman S, et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. Oct 2021;23(10):1793-1806. doi:10.1038/s41436-021-01203-z
61. ACOG. Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstetrics and gynecology. Mar 2017;129(3):e41-e55. doi:10.1097/aog.0000000000001952
62. Wilson, Bie, Armour, et al. Joint SOGC–CCMG Opinion for Reproductive Genetic Carrier Screening: An Update for All Canadian Providers of Maternity and Reproductive Healthcare in the Era of Direct-to-Consumer Testing. Journal of Obstetrics and Gynaecology Canada. 2016;38(8):742–762.
63. Audibert F, De Bie I, Johnson J-A, et al. No. 348-Joint SOGC-CCMG Guideline: Update on Prenatal Screening for Fetal Aneuploidy, Fetal Anomalies, and Adverse Pregnancy Outcomes. Journal of Obstetrics and Gynaecology Canada. 2017/09/01/ 2017;39(9):805-817. doi:10.1016/j.jogc.2017.01.032
64. Armour CM, Dougan SD, Brock JA, et al. Practice guideline: joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J Med Genet. Apr 2018;55(4):215-221. doi:10.1136/jmedgenet-2017-105013
65. Carter MT, Srour M, Au PB, et al. Genetic and metabolic investigations for neurodevelopmental disorders: position statement of the Canadian College of Medical Geneticists (CCMG). J Med Genet. Jun 2023;60(6):523-532. doi:10.1136/jmg-2022-108962
66. Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. European journal of human genetics : EJHG. Apr 2015;23(4):417-25. doi:10.1038/ejhg.2014.185
Coding Section
| Codes | Number | Description |
| CPT | 81243 | FMR1 (fragile X mental retardation 1) (e.g., fragile X mental retardation) gene analysis; evaluation to detect abnormal (e.g., expanded) alleles |
| 81244 | FMR1 (fragile X mental retardation 1) (e.g., fragile X mental retardation) gene analysis; characterization of alleles (e.g., expanded size and methylation status) |
|
| 88248 | Chromosome analysis for breakage syndromes; baseline breakage, score 50 – 100 cells, count 20 cells, 2 karyotypes (e.g., for ataxia telangiectasia, Fanconi anemia, fragile X) |
|
| 96040 | Medical genetics and genetic couseling services, each 30 minutes face-to-face with patient/family |
|
| S0265 | Genetic counseling, under physician supervisor, each 15 minutes |
|
| Type of Service |
||
| Place of Service |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 01/23/2026 | Annual review, no change to policy intent. Updating policy for clarity and consistency, table of terminology, rationale, and references. |
| 01/23/2025 | Annual review, no change to policy intent. Updating description, rationale, references. |
| 01/24/2024 | Annual review, updating policy verbiage to add coverage of AGG interruption testing. Also updating rationale and references. |
| 01/25/2023 | Annual review, no change to policy intent. Policy verbiage updated for clarity. Also updating description, rationle and references. |
| 08/12/2022 | Interim review. Adding statement regarding testing more than once per lifetime and updating verbiage 2e to state fetuses and offspring instead off offspring alone. Also updating description, rationale and references. |
| 01/25/2022 |
Annual review, no change to policy intent. Updating policy number, rationale and references. Adding CPT codes 96040 and S0265. |
| 01/06/2021 |
Annual review, no change to policy intent. Updating description, rationale and references. |
| 01/06/2020 |
Annual review, no change to policy intent. Updating coding. |
| 06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made. |
| 01/02/2019 |
Annual review, no change to policy intent. |
| 12/19/2018 |
Updating with 2019 codes. |
| 01/31/2018 |
Annual review, reformatting policy verbiage for clarity. Also updating background, description, guidelines, rationale and references. |
| 04/26/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
| 09/27/2016 |
Updated the word guideline to policy when applicable. No change to policy intent. |
| 07/07/2016 |
Annual review, no change to policy intent. Updating background, description, regulatory status, rationale, references and appendix 1. |
| 07/21/2015 |
Annual review, no change to policy intent. Updated background, description, guidelines, rationale and references. Added coding and appendix 1 |
| 07/29/2014 |
Annual review. Added related policy. Updated desciption, background, rationale and references. Updated policy verbiage to include that uses other than those discussed are investigational. |