
Genetic Testing of Mitochondrial Disorders - CAM 258
Description
Mitochondrial disease refers to a heterogeneous group of disorders caused by dysfunctional mitochondria; the organelles responsible for oxidative phosphorylation within the cell. Mitochondrial diseases are classified according to the primary genetic defect, including those affecting respiratory chain proteins or ancillary proteins, mitochondrial RNA translation defects, inner membrane lipid defects, mutations causing depletion of mitochondrial DNA, or mutations to mitochondrial dynamics. Tissues with high energy demands, such as brain, heart, and skeletal muscle, are most affected by mitochondrial diseases.1 As a result, mitochondrial encephalopathy and cardiomyopathy are the most prominent manifestations.2 Some examples of mitochondrial disorders include mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS), myoclonic epilepsy and ragged‐red‐fibers (MERRF), chronic progressive external ophthalmoplegia (CPEO), Kearns‐Sayre syndrome (KSS), and maternally inherited Leigh syndrome (MILS).3
When pursuing genetic testing for mitochondrial disorders, genetic counseling is strongly recommended.
Regulatory Status
A search of the FDA database on 10/27/2020 using the terms “mtDNA” and “mitochondrial disease” yielded 0 results. Many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- As an alternative to muscle biopsy for individuals with clinical signs and symptoms consistent with a specific mitochondrial disorder, but for whom a definitive diagnosis cannot be made without genetic testing, genetic testing to confirm the diagnosis of a mitochondrial disorder is considered MEDICALLY NECESSARY.
- For individuals who are strongly suspected of having a mitochondrial disorder but who do not have symptomology associated with a specific mitochondrial condition, genomic sequencing and large deletion detection of the entire mitochondrial genome, with heteroplasmy detection, is considered MEDICALLY NECESSARY.
- For individuals who are strongly suspected of having a mitochondrial disorder but who do not have symptomology associated with a specific mitochondrial condition, genetic testing for nuclear encoded mitochondrial genes (in conjunction with or after testing for variants in the mitochondrial genome) is considered MEDICALLY NECESSARY.
- For the diagnosis of mitochondrial DNA (mtDNA) depletion syndromes, quantification of mtDNA in tissue is considered MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- The combination of mitochondrial genome testing with nuclear genome testing with whole genome sequencing (e.g., Genomic Unity ® Whole Genome Analysis), is considered NOT MEDICALLY NECESSARY.
- For all other situations not discussed above, genetic testing for mitochondrial disorders is considered NOT MEDICALLY NECESSARY.
Table of Terminology
| Term |
Definition |
| ACGS |
Association for Clinical Genomic Science |
| BGS |
Bigenomic sequencing |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CM |
Cardiomyopathy |
| CMS |
Centers for Medicare & Medicaid Services |
| COX10 |
|
| CPEO |
Chronic progressive external ophthalmoplegia |
| DNA |
Deoxyribonucleic acid |
| dThd |
Crystalline nucleoside of thymine |
| EAN |
European Academy of Neurology |
| ES |
Exome sequencing |
| FDA |
Food and Drug Administration |
| GLUL |
Glutamine synthetase |
| KSS |
Kearns‐Sayre syndrome |
| LDT |
Laboratory-developed test |
| LHON |
Leber hereditary optic neuropathy |
| LR-PCR |
Long range polymerase chain reaction |
| MD |
Mitochondrial disease |
| MELAS |
Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes |
| MERRF |
Myoclonic epilepsy and ragged‐red‐fibers |
| MILS |
Maternally inherited Leigh syndrome |
| MNGIE |
Mitochondrial neurogastrointestinal encephalomyopathy |
| MPS |
Massively parallel sequencing |
| MRPS23 |
Mitochondrial ribosomal protein S23 |
| mtDNA |
Mitochondrial deoxyribose nucleic acid |
| MT-RNR1 |
Mitochondrially encoded 12S ribosomal RNA |
| NADH |
Nicotinamide adenine dinucleotide |
| NAMDC |
North American Mitochondrial Disease Consortium |
| NARP |
Neuropathy, ataxia, and retinitis pigmentosa |
| nDNA |
Nuclear deoxyribose nucleic acid |
| NGS |
Next-generation sequencing |
| nWES |
Nuclear whole exome sequencing |
| ONT |
Oxford Nanopore Technologies |
| PEO |
Progressive external ophthalmoplegia |
| PGT |
Preimplantation genetic testing |
| PMD |
Primary mitochondrial disease |
| PNPLA4 |
Patatin like phospholipase domain containing 4 |
| POLG |
Polymerase gamma |
| POLG1 |
Deoxyribose nucleic acid polymerase subunit gamma |
| Q10 |
Coenzyme Q10 (also known as ubiquinone) |
| QRSL1 |
Glutaminyl-trna synthase (glutamine-hydrolyzing)-like 1 |
| RMND1 |
Required for meiotic nuclear division protein 1 |
| RNA |
Ribonucleic acid |
| TP |
Thymidine phosphorylase |
| VUS |
Variant of uncertain significance |
| WES |
Whole exome sequencing |
| WGS |
Whole genome sequencing |
Rationale
MMitochondrial diseases are generally inherited and present with an elaborate genetic makeup.4 Several different types of mitochondrial diseases and resulting ailments exist which include “mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS), myoclonic epilepsy and ragged‐red‐fibers (MERRF), CPEO, Kearns‐Sayre syndrome (KSS), neuropathy, ataxia, and retinitis pigmentosa (NARP), maternally inherited Leigh syndrome (MILS), and Leber hereditary optic neuropathy (LHON).”3 These diseases can be caused by mutations in either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA); many different mitochondrial disease mutations have been identified, but clinical symptoms are extremely variable and may even differ between patients carrying the same mtDNA mutation.3 Symptoms present in several areas of the body, including the central nervous system, cardiovascular system, gastrointestinal system, endocrine system, and neuromuscular system.3 Clinical differences depend on age of onset, affected organ or tissue, and disease progression. Phenotypic variability and severity can create challenges when diagnosing affected patients.1
Pathogenic variants in more than 300 genes have been associated with mitochondria-related disorders.5 For example, infantile onset of mitochondrial diseases with multiple mitochondrial respiratory chain defects results from mutation(s) in the Required for Meiotic Nuclear Division protein 1 (RMND1) gene.6 Further, a pathogenic variant in the ubiquinone biosynthesis protein (COQ4) gene has been affiliated with mitochondrial disease development.4,7 Patients with mutations in the NADH dehydrogenase mitochondrial (MT-ND1 and NDUF) genes share phenotypic and genotypic correlations in Leigh syndrome.8 The individual symptoms are nonspecific, and symptom patterns can overlap considerably. As a result, a patient often cannot be easily classified into one particular syndrome.9
Previously, the prevalence of mitochondrial diseases was considered low. With the advent of advanced genetic analysis tools, numerous studies now report a higher incidence of mitochondrial-associated mutations. A meta-analysis of the prevalence of the three primary mtDNA mutations that cause LHON in Europe shows a prevalence of approximately 1:45,000.10 A longitudinal study in Sweden reports an incidence of mitochondrial encephalomyopathies, in general, at 1:11,000, and an incidence of infantile mitochondrial myopathy with cytochrome C oxidase deficiency at 1:51,000. The authors conclude that “mitochondrial encephalomyopathies are relatively common neurometabolic disorders in childhood.”11 A 2015 study in the United Kingdom reports that “the total prevalence of adult mitochondrial disease, including pathogenic mutations of both the mitochondrial and nuclear genomes (≈1 in 4,300), is among the commonest adult forms of inherited neurological disorders.”12 An Australian study estimates a “minimum birth prevalence of 13.1/100 000 or 1/7634 for respiratory chain disorders with onset at any age.”13
Barca, et al. (2020) evaluated the phenotypic and molecular characteristic of 666 participants in the North American Mitochondrial Disease Consortium (NAMDC) aiming to better understand mitochondrial diseases in North America. Multisystemic disorder was the most common diagnosis (113 participants), compared to classical mitochondrial syndrome. Leigh syndrome (97 participants) and MELAS (71 participants) were the most frequent classical syndromes. Pathogenic variants in mtDNA, with the most common variants being POLG1 and PDHA1, were more common than pathogenic gene variants.14
The figure below (taken from O'Brien, et al. (2014)) gives examples of classical phenotype mitochondrial diseases along with the clinical features and molecular genetics associated with each disorder.
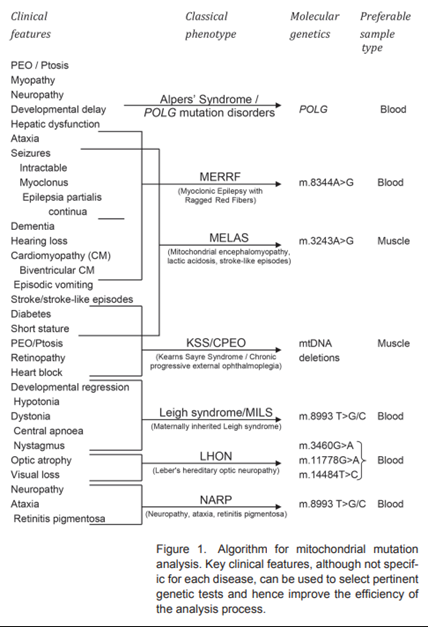
The evaluation and diagnostic approach for a mitochondrial disorder varies according to age, clinical phenotype, and presumed inheritance pattern. Biochemical testing is indicated for patients who do not have a clear clinical picture of one specific disorder. Measurement of serum lactic acid is often used as a screening test, but the test is neither sensitive nor specific for mitochondrial disorders.16 “Identifying causative mutations underlying mitochondrial dysfunction is the ultimate gold standard for the diagnosis. Two mitochondrial diseases (MNGIE and coenzyme Q10 deficiency) are particularly important to identify because of potential treatments.”1
Neuhofer and Prokisch (2024) explored digenic inheritance—the interplay between mitochondrial and nuclear gene mutations—in mitochondrial disease expression. The authors presents cases where mutations in both mtDNA (e.g., m.11778G>A) and nuclear genes (e.g., DNAJC30, NDUFS2, NDUFS8) result in overlapping Leigh syndrome and LHON phenotypes. Additional examples include combinations such as VARS2/MT-TV and YARS/MT-ND1, which modify disease penetrance and severity. The findings challenge the one-gene-one-disease model, suggesting multigene interactions contribute to phenotypic variability and should inform next-generation sequencing analyses and variant interpretation.17
Proprietary Testing
Vairantyx provides two separate tests, Exome Plus Analysis and Whole Genome Analysis. While the manufacturer offers two tests, the descriptions of what these two tests are reporting on is nearly identical and, currently, it is unclear as to the difference between them.
Genomic Unity ® Exome Plus Analysis
This test provides a full, phenotypically driven analysis of all relevant genes and variant types. This analysis includes genome-wide sequence analysis, genome-wide structural variant analysis, mitochondrial genome analysis with heteroplasmy (≥ five percent), mitochondrial genome large deletions analysis, short tandem repeat (STR) analysis (AFF2, AR, ARX, ATN1, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, ATXN10, C9ORF72, CACNA1A, CNBP, CSTB,- DIP2B, DMPK, FGF14, FMR1, FOXL2, FXN, GIPC1, GLS, HTT, JPH3, LRP12, NOP56, NOTCH2NLC, PABPN1, PHOX2B, PPP2R2B, RFC1, TBP, TCF4, VWA1, ZFHX3, ZIC2). This test has a performance rate of >99.9% sensitivity, >99.9% specificity, 99.8% positive predictive value, and a 96% clinical sensitivity for structural variants.18
Genomic Unity ® Whole Genome Analysis
Whole Genome Analysis can be ordered as a first-line test or when previous testing was non-diagnostic. This type of analysis includes genome-wide sequence analysis, genome-wide structural variant analysis, mitochondrial genome sequence analysis with heteroplasmy (≥ five percent) and large deletion analysis, STR analysis for adult-onset movement disorder with or without cognitive involvement (AFF2, AR, ARX, ATN1, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, ATXN10, C9ORF72, CACNA1A, CNBP, CSTB, DIP2B, DMPK, FGF14, FMR1, FOXL2, FXN, GIPC1, GLS, HTT, JPH3, LRP12, NOP56, NOTCH2NLC, PABPN1, PHOX2B, PPP2R2B, RFC1, SOX3, TBP, TCF4, VWA1, ZFHX3, ZIC2). This test has a performance rate of >99.9% sensitivity, >99.9% specificity, >99.8% positive predictive value, and a 96% clinical sensitivity for structural variants.19
Clinical Utility and Validity
Clinical utility is high for confirming the diagnosis of mitochondrial disorders in people who have clinical features consistent with a specific mitochondrial disease. In these patients, a positive result on genetic testing can avoid a muscle biopsy and eliminate the need for further clinical workup.20 Additionally, genetic testing may impact reproductive decision making when a defined mitochondrial disease is present in the family that is severe enough to cause impairment and/or disability. If genetic testing is used in this situation, there will be a decreased risk of a mitochondrial disorder in the offspring. Such testing includes whole exome sequencing, whole genome sequencing, and whole mitochondrial sequencing.
The most common first-line diagnostic test for a mitochondrial disease is massively parallel sequencing (MPS), also known as next-generation sequencing (NGS); other accepted methods included targeted panels, whole exome sequencing (WES) and whole genome sequencing (WGS).21 Broad-based exome sequencing has been considered the first-line diagnostic tool for primary mitochondrial disease (PMD),22 and other researchers have used WES in tandem with rapid mitochondrial genome (mtDNA) sequencing for diagnoses.23
Whole exome sequencing is likely to increase the detection rate but will also increase the rate of identifying a variant of uncertain significance (VUS). In one study from the U.K., 53 patients who had biochemical evidence of a mitochondrial disorder but were negative on genetic testing of the primary mitochondrial disorder mutations, underwent whole exome sequencing. Probable pathogenic mutations causative of a mitochondrial disorder was identified in 28 patients (53%), and an additional four patients had variants that were possibly pathogenic.24 “False negative rates vary by genomic region; therefore, genomic testing may not be as accurate as targeted single gene testing or multigene molecular genetic testing panels. Most laboratories confirm positive results using a second, well-established method.”9
Expanded panels are defined as panels that include more genes than are associated with any specific disorder. When these panels are used in individuals with nonspecific signs and symptoms that are not consistent with a “classic” presentation of a mitochondrial disorder, the probability of finding a pathogenic mutation is considerably less. Conversely, the likelihood of a false-positive result and the number of VUS may be substantially increased.15 Table 1, below, lists examples of commercially available expanded genetic panels for mitochondrial disorders.
Table 1. Examples of Commercially Available Expanded Genetic Panels for Mitochondrial Disorder
| Laboratory |
Lab Test or Panel |
Number of Genes Included on Panel |
| Gene Dx® (Gaithersburg, MD) |
MitoXpanded Panel25 |
~1800 |
| Combined Mito Genome Plus Mito Focused Nuclear Gene Panel26 |
202 |
|
| ARUP® (Salt Lake City, UT) |
Mitochondrial Disorders Panel27 |
>150 |
| Baylor® Genetics (Houston, TX) |
Mitome200 Nuclear Panel28 |
164 |
| Medical Neurogenetics® (Atlanta, GA) |
Mitochondrial Genome Sequencing & Deletion Analysis29 |
>300 |
WWood, et al. (2019) compared the mtDNA sequencing results of the long-read sequencing technology Oxford Nanopore Technologies (ONT) MinION to the Illumina MiSeq, a NGS platform. A total of 12 patients participated in this study (three healthy controls and nine with known mtDNA deletion disorders). Both of these NGS methods were more efficient than LR-PCR and/or Southern Blotting; further, MinION proved to be just as accurate as the Illumina MiSeq in this study, successfully identifying all mtDNA deletions.30 This tool may assist in making the mitochondrial disease diagnostic process more efficient and cost effective in the future.
Wagner, et al. (2019) researched the effectiveness of exome sequencing (ES) for mitochondrial disease diagnostics; data was used from 2111 clinical cases. The researchers stated that “ES identified known pathogenic mtDNA point mutations in 38 individuals, increasing the diagnostic yield by nearly two percent. Analysis of mtDNA variants by ES had a high recall rate (96.2 ± 5.6%) and excellent precision (99.5 ± 2.2%) when compared to the gold standard of targeted mtDNA next-generation sequencing.”31 These results suggest that ES should be considered as a diagnostic tool for both nDNA and mtDNA testing.
Legati, et al. (2016) suggest a two-tiered approach to genetic testing where targeted NGS is used first in cases of suspected mitochondrial disorders followed by WES in patients who have inconclusive results. “Importantly, WES on selected cases has unraveled the presence of pathogenic mutations in genes encoding non-mitochondrial proteins (e.g. the transcription factor E4F1), an observation that further expands the intricate genetics of mitochondrial disease and suggests a new area of investigation in mitochondrial medicine.”32
Pronicka, et al. (2016) suggest that WES is a better diagnostic tool than NGS. A total of 113 pediatric Polish patients participated in this study; variants were identified in both nDNA and mtDNA.33 WES was able to identify “likely causative mutations” in 67 patients, with 50.5% of all detected genetic changes novel variants; further, “In 47 patients, changes in 31 MD-related genes (ACAD9, ADCK3, AIFM1, CLPB, COX10, DLD, EARS2, FBXL4, MTATP6, MTFMT, MTND1, MTND3, MTND5, NAXE, NDUFS6, NDUFS7, NDUFV1, OPA1, PARS2, PC, PDHA1, POLG, RARS2, RRM2B, SCO2, SERAC1, SLC19A3, SLC25A12, TAZ, TMEM126B, VARS2) were identified.”33
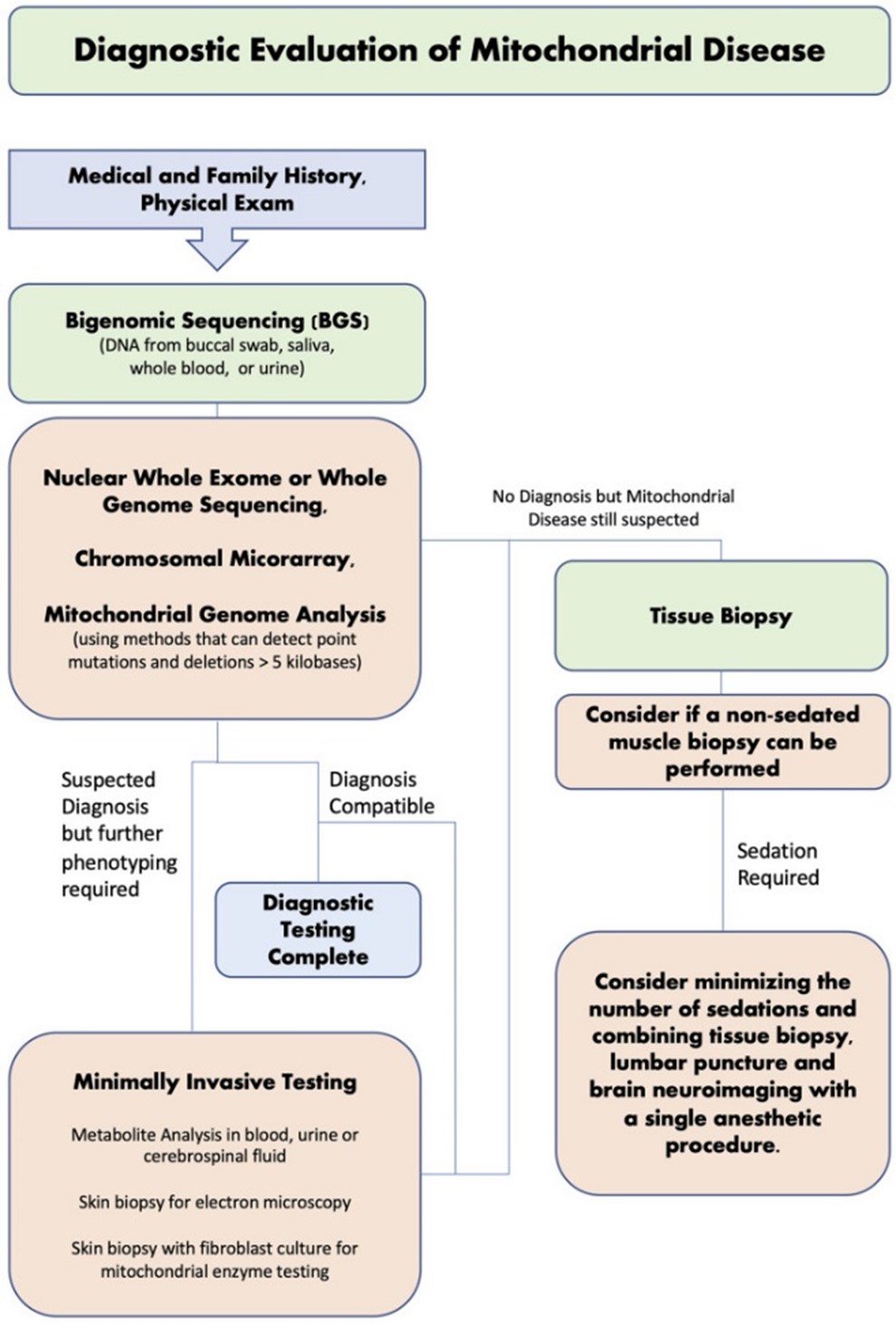
In a study by Kerr, et al. (2020), 390 patients were recruited and tested for mitochondrial disease (MD) through traditional diagnostic pathways (metabolite analysis, tissue neuropathology and respiratory chain enzyme activity) and new diagnostic approaches (NGS and nuclear WES (nWES)). Testing through traditional diagnostic methods resulted in a mitochondrial diagnosis in 115 out of 390 patients (29.50%). In comparison, 116 out of 390 patients were recruited for NGS, which identified 36 patients (31%) with a mitochondrial diagnosis. To confirm diagnosis, patients were further tested through nWES, which provided a secondary diagnosis in “two cases who already had a pathogenic variant in mtDNA, and a revised diagnosis (GLUL) in one case that had abnormal pathology but no pathogenic mtDNA variant.”34 nWES also identified a mitochondrial diagnosis in one patient who tested negative from NGS. The author offers a diagnostic evaluation strategy, as shown in the figure below, and recommends that “a non-invasive, bigenomic sequencing (BGS) approach (using both a nWES and optimized mtDNA analysis to include large deletions) should be the first step in investigating for MDs. There may still be a role for tissue biopsy in unsolved cases or when the diagnosis is still not clear after NGS studies.”34
Another study clearly demonstrates the heterogeneity of genetic mutations causing mitochondrial disorders. Of the 142 patients with childhood-onset mitochondrial disorders, researchers “identified 37 novel mutations in known mitochondrial disease genes and three mitochondria-related genes (MRPS23, QRSL1, and PNPLA4) as novel causative genes.”35 These researchers utilized whole mtDNA and exome and chromosomal aberration analysis approaches to “enhance the ability to identify pathogenic gene mutations in patients.”35A study by Fang, et al. (2017) recruited 141 children with suspected mitochondrial disorders and used NGS to identify genetic characteristics. Forty children were gene confirmed with a known mitochondrial disease; 62.5% of those cases were due to a mtDNA mutation and 37.5% due to a nDNA mutation. This study found the most prevalent disorders to be Leigh syndrome and MELAS.36
Spath, et al. (2021) studied parallel preimplantation genetic testing (PGT) for mtDNA disease and aneuploidy on four patients at risk of transmitting mtDNA disease. Aneuploidy is the condition of having an abnormal number of chromosomes. “Half of the embryos tested were shown to be aneuploid (16/33).” Notably, not all the participants had the same mtDNA mutations. Patients A and D had a m.8993T>G mutation; mutations were detected in embryo biopsies from Patient A but not Patient B. Patient C had a m.10191T.G mutation, no mutations were detected in embryo biopsies from Patient C. Patient D had a m3243A>G mutation, mutations were detected in embryo biopsies from Patient D. Patients A and D displayed somatic heteroplasmy for mtDNA mutations, “heteroplasmic women had a higher incidence of affected embryos than those with undetectable somatic mutant mtDNA but were still able to produce mutation-free embryos.” The authors concluded that “strategies providing a combination of PGT for mtDNA disease and aneuploidy may be advantageous compared with approaches that consider only mtDNA.”37
Wu, et al. (2021) evaluated the cost-efficiency and cost-benefit of genomic sequencing for children presenting clinical indications of mitochondrial diseases compared to the conventional diagnosis pathway in Australia. The authors used a decision tree model approach and a discrete-event simulation approach on data from 78 pediatric-onset patients. The conventional diagnosis pathway refers to a diagnostic workup including metabolic, imaging, and histopathological investigations, and genetic testing. Genomic sequencing was less costly and more effective than conventional care. The authors concluded that “genomic sequencing is cost-saving relative to traditional investigative approaches, while enabling more diagnoses to be made in a timely manner, offering substantial personal benefits to children and their families.”38
Farahvash, et al. (2021) did a case study on two siblings with mitochondrial neurogastrointestinal encephalopathy disease (MNGIE). The two patients experienced gastrointestinal and cachexia symptoms for years and underwent numerous exploratory tests and surgeries which did not improve their condition. Eventually, the patients were diagnosed with MNGIE following genetic testing for the TYMP gene. The authors concluded that “MNGIE diagnosis is important to establish to avoid unnecessary invasive testing for gastrointestinal, ophthalmological, and neurological symptoms and to ensure patients receive appropriate nutritional and genetic counselling.”39
Future Therapies and Directions
Wen, et al. (2025) conducted a review discussing the emerging pharmacological treatments for mitochondrial disorders, emphasizing how advancements in drug technology are targeting mitochondrial dysfunction. Therapeutics such as CoQ10, idebenone, EPI-743, elamipretide, and IFN- γ aim to improve energy production, reduce oxidative stress, and restore mitochondrial integrity. CoQ10 enhances ATP production and stabilizes mitochondrial membranes. Idebenone, a CoQ10 analog, bypasses complex I dysfunction to maintain cellular energy in conditions like LHON and MELAS. Other agents such as erythropoietin derivatives and IFN- γ are being explored for Friedreich’s ataxia, reflecting a broader shift toward targeted, mechanism-based therapies for mitochondrial diseases.40
Mitochondrial Medicine Society
The Mitochondrial Medicine Society published the following consensus recommendations on genetic testing for mitochondrial disorders:41
- “Massively parallel sequencing/NGS of the mtDNA genome is the preferred methodology when testing mtDNA and should be performed in cases of suspected mitochondrial disease instead of testing for a limited number of pathogenic point mutations.
- Patients with a strong likelihood of mitochondrial disease because of a mtDNA mutation and negative testing in blood, should have mtDNA assessed in another tissue to avoid the possibility of missing tissue-specific mutations or low levels of heteroplasmy in blood; tissue-based testing also helps assess the risk of other organ involvement and heterogeneity in family members and to guide genetic counseling.
- Heteroplasmy analysis in urine can selectively be more informative and accurate than testing in blood alone, especially in cases of MELAS due to the common m. 3243A>G mutation.
- mtDNA deletion and duplication testing should be performed in cases of suspected mitochondrial disease via NGS of the mtDNA genome, especially in all patients undergoing a diagnostic tissue biopsy.
- If a single small deletion is identified using polymerase chain reaction–based analysis, then one should be cautious in associating these findings with a primary mitochondrial disorder.
- When multiple mtDNA deletions are noted, sequencing of nuclear genes involved in mtDNA biosynthesis is recommended.
- When a tissue specimen is obtained for mitochondrial studies, mtDNA content (copy number) testing via real-time quantitative polymerase chain reaction should strongly be considered for mtDNA depletion analysis because mtDNA depletion may not be detected in blood.
- mtDNA proliferation is a nonspecific compensatory finding that can be seen in primary mitochondrial disease, secondary mitochondrial dysfunction, myopathy, hypotonia, and as a by-product of regular, intense exercise.
- When considering nuclear gene testing in patients with likely primary mitochondrial disease, NGS methodologies providing complete coverage of known mitochondrial disease genes is preferred. Single-gene testing should usually be avoided because mutations in different genes can produce the same phenotype. If no known mutation is identified via known NGS gene panels, then whole exome sequencing should be considered.”
The Mitochondrial Medicine Society also commented on the set of mtDNA depletion syndromes, which are “characterized by a significant reduction in mtDNA copy number in affected tissues.” They note that diagnosis of these conditions “requires quantification of mtDNA content, typically in affected tissue, with identification of a significant decrease below the mean of normal age, gender, and tissue-specific control when normalized to nDNA tissue content.” Since NGS of the mtDNA genome does not identify mtDNA content, a separate quantitative real-time polymerase chain reaction must be used.41
The Mitochondrial Medicine Society in 2017 released their guidelines regarding patient care standards. Within this set of guidelines, they state, “Pregnancy in mitochondrial disease also elicits the concern of transmission of a genetic disorder. Appropriate preconception genetic counseling and discussion of options of prenatal testing are needed. A fetus affected by mitochondrial disease may also be at higher risk for prenatal morbidity. Finally, premature ovarian failure is a feature of several mitochondrial disorders and affected women should be referred for assisted reproductive technologies if they wish to have children.”42
Association for Clinical Genomic Science (ACGS)
The ACGS published guidelines for the genetic testing strategies for diagnostic and familial testing, variant interpretation and reporting, and prenatal diagnosis and reproductive options. The guidelines report the minimal level of testing recommended for the most common diagnostic referrals, listed below, noting possible further testing with nuclear DNA testing and whole mtDNA sequencing for all phenotypes.
- Phenotype: ataxia, possible diagnosis of late-childhood or adult-onset peripheral neuropathy, pigmentary retinopathy (NARP) or mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Minimum level of testing for: m.8993T>G p.(Leu156Arg), m.8993T>C p.(Leu156Pro)(MT-ATP6), m.3243A>G (MT-TL1).
- Phenotype: cardiomyopathy, familial hypertrophic cardiomyopathy with maternal inheritance. Minimum level of testing for: m.4300A>G (MT-TI), m.3243A>G (MT-TL1).
- Phenotype: diabetes mellitus and sensorineural hearing loss. Minimum level of testing for: m.3243A>G (MT-TL1), m.3243A>G (MT-TL1).
- Phenotype: encephalopathy/seizures with lactic acidosis, possible diagnosis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) or infantile onset subacute relapsing encephalopathy, cerebellar and brain stem signs (MILS). Minimum level of testing for: m.3243A>G, (MT-TL1), m.8993T>G p.(Leu156Arg), m.8993T>C p.(Leu156Pro), (MT-ATP6).
- Phenotype: non-syndromic sensorineural hearing loss, particularly if onset following aminoglycoside exposure. Minimum level of testing for m.1555A>G (MT-RNR1).
- Phenotype: Kearns-Sayre syndrome onset, below the age of 20 years, PEO and pigmentary retinopathy with one of either cardiac conduction block, cerebrospinal fluid protein concentration greater than 100 mg/dL, or cerebellar ataxia. Minimum level of testing for: large-scale mtDNA rearrangements (single and multiple deletions), m.3243A>G (MT-TL1).
- Phenotype: Leber hereditary optic neuropathy, optic atrophy, childhood or midlife (adult-onset) acute or subacute painless bilateral central vision loss. Minimum level of testing for: m.3460G>A p.(Ala52Thr) (MT-ND1), m.11778G>A p.(Arg340His) (MT-ND4), m.14484T>C p.(Met64Val) (MT-ND6).
- Phenotype: mtDNA depletion syndrome, neonatal or infantile hepatocerebral, myopathic, encephalomyopathic or neurogastrointestinal presentations; may also include growth failure, lactic acidosis, and hypoglycemia. Minimum level of testing for: mtDNA copy number analysis.
- Phenotype: myoclonic epilepsy, myoclonus, seizures; cerebellar ataxia; myopathy. Minimum level of testing for: m.8344A>G (MT-TK), m.3243A>G (MT-TL1).
- Phenotype: Pearson syndrome, sideroblastic anemia of childhood; Pancytopenia; Exocrine pancreatic failure. Minimum level of testing for: large-scale mtDNA rearrangements.
- Phenotype: progressive external ophthalmoplegia (PEO), ptosis, typically adult-onset ptosis, paralysis of the extraocular muscles (ophthalmoplegia), oropharyngeal weakness, and variably severe proximal limb weakness. Minimum level of testing for: large-scale mtDNA rearrangements (single and multiple deletions, m.3243A>G (MT-TL1).
- Phenotype: stroke-like episodes, typically before age 40 years. Minimum level of testing for: m.3243A>G (MT-TL1).43
European Academy of Neurology (EAN)
Regarding genetic testing for mitochondrial encephalopathy, lactic acidosis and stroke‐like episodes (MELAS), the EAN recommends “Urgent genetic testing for MELAS should be considered in patients presenting with suspected SLE [stroke‐like episode]. Search for the m.3243A>G variant (in urine where possible) and, if m.3243A>G variant is negative, POLG sequencing is required. Muscle biopsy should be considered after excluding m.3243A>G and POLG variants.” The EAN also notes that valproic acid is “contraindicated, mainly in patients with POLG variants.”44
MNGIE International Network
The MNGIE [Mitochondrial neurogastrointestinal encephalomyopathy] International Network recommended TYMP sequencing to confirm a MNGIE diagnosis. If a variant of unknown significance or a wild-type variant is found, the Network recommends biochemical testing of thymidine (dThd) and deoxyuridine levels to determine thymidine phosphorylase (TP) activity. The Network also remarks on several treatment options (both short- and long-term), which focus on restoring the biochemical balance of the patient.45
United Kingdom Best Practice Guidelines
In 2024, a group of clinical scientists part of the “United Kingdom National Health Service Highly Specialised Services for Rare Mitochondrial Disorders” group revised and updated their 2008 guidelines for genetic testing for mitochondrial disease.46
In a proband with suspected mitochondrial disease, the group recommends that “first-line targeted testing of blood DNA can be appropriate for routine referrals (where there is not an urgent clinical need to obtain a diagnosis), for clinical presentations which are highly suggestive of a particular variant or gene (e.g., MELAS, MERRF, LHON, Pearson syndrome, POLG-related disorders), and/or where resources are limited. In patients where no pathogenic variant is detected in blood, additional clinical information and samples (e.g., urine) or a muscle biopsy for biochemical and histopathological investigations may be suggested.” The group also notes that “alternatively, comprehensive NGS-based testing can be adopted for sensitive and efficient first-line genetic testing. This approach can be followed for all referral indications but is particularly appropriate for more complex phenotypes and/or for urgent referrals.”46
The group notes that “specific single gene testing remains a valuable simple and rapid first-line test when the associated disorder is common, for treatable disorders with a characteristic phenotype, when there are common founder variants(s), and/or if biochemical evidence points to a defect in the particular gene.”46
For family testing, the group recommends that “following a genetic diagnosis of mitochondrial disease, it is recommended that the proband (and their family) are referred to clinical genetics or a specialist mitochondrial service for appropriate genetic counselling and family follow-up.” The group notes that testing of asymptomatic children below the age of 16 with a family history of mtDNA-related mitochondrial disease is challenging, and recommends that “laboratories discuss referrals for genetic testing of asymptomatic children with specialised clinicians or clinical geneticists wherever possible prior to testing.”46
References
- O'Ferrall E. Mitochondrial myopathies: Clinical features and diagnosis. Wolters Kluwer. Updated Mar 24, 2025. https://www.uptodate.com/contents/mitochondrial-myopathies-clinical-features-and-diagnosis
- Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J. 2013;40(4):385-94.
- Hatakeyama H, Goto Y. Concise Review: Heteroplasmic Mitochondrial DNA Mutations and Mitochondrial Diseases: Toward iPSC-Based Disease Modeling, Drug Discovery, and Regenerative Therapeutics. Stem Cells. Apr 2016;34(4):801-8. doi:10.1002/stem.2292
- Lightowlers RN, Taylor RW, Turnbull DM. Mutations causing mitochondrial disease: What is new and what challenges remain? Science. Sep 25 2015;349(6255):1494-9. doi:10.1126/science.aac7516
- Thompson K, Collier J, Glasglow R, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. 2019;doi:10.1002/jimd.12104
- Ng YS, Alston CL, Diodato D, et al. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J Med Genet. Nov 2016;53(11):768-775. doi:10.1136/jmedgenet-2016-103910
- Ling T-k, Law C-y, Ko C-h, et al. A common COQ4 mutation in undiagnosed mitochondrial disease: a local case series. 2019;51:S112–S113. doi:10.1016/j.pathol.2018.12.317
- Sofou K, de Coo IFM, Ostergaard E, et al. Phenotype-genotype correlations in Leigh syndrome: new insights from a multicentre study of 96 patients. J Med Genet. Jan 2018;55(1):21-27. doi:10.1136/jmedgenet-2017-104891
- Chinnery PF. Mitochondrial Disorders Overview. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews((R)). University of Washington, Seattle; 2014.
- Mascialino B, Leinonen M, Meier T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. European journal of ophthalmology. May-Jun 2012;22(3):461-5. doi:10.5301/ejo.5000055
- Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Annals of neurology. Mar 2001;49(3):377-83.
- Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of neurology. May 2015;77(5):753-9. doi:10.1002/ana.24362
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain : a journal of neurology. Aug 2003;126(Pt 8):1905-12. doi:10.1093/brain/awg170
- Barca E, Long Y, Cooley V, et al. Mitochondrial diseases in North America: An analysis of the NAMDC Registry. Neurol Genet. Apr 2020;6(2):e402. doi:10.1212/nxg.0000000000000402
- O'Brien M, Cryan J, Brett F, Howley R, Farrell M. Ten years on: genetic screening for mitochondrial disease in Ireland. Clinical neuropathology. Jul-Aug 2014;33(4):279-83. doi:10.5414/np300722
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nature reviews Genetics. Dec 2012;13(12):878-90. doi:10.1038/nrg3275
- Neuhofer CM, Prokisch H. Digenic Inheritance in Rare Disorders and Mitochondrial Disease-Crossing the Frontier to a More Comprehensive Understanding of Etiology. Int J Mol Sci. Apr 23 2024;25(9)doi:10.3390/ijms25094602
- Variantyx. Genomic Unity® Exome Plus Analysis. https://www.variantyx.com/products-services/rare-disorder-genetics/comprehensive-analyses/genomic-unity-exome-plus-analysis/
- Variantyx. Genomic Unity® Whole Genome Analysis. https://www.variantyx.com/products-services/rare-disorder-genetics/comprehensive-analyses/genomic-unity-whole-genome-analysis/
- Dimmock DP, Lawlor MW. Presentation and Diagnostic Evaluation of Mitochondrial Disease. Pediatr Clin North Am. Feb 2017;64(1):161-171. doi:10.1016/j.pcl.2016.08.011
- Horvath R, Chinnery P. Diagnostic Approach to Mitochondrial Diseases. 2019.
- McCormick EM, Zolkipli-Cunningham Z, Falk MJ. Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr. Dec 2018;30(6):714-724. doi:10.1097/mop.0000000000000686
- Akesson L, Eggers S, Chong B, et al. Rapid mitochondrial genome (MTDNA) sequencing: facilitating rapid diagnosis of mitochondrial diseases in paediatric acute care. 2019;51(1):118–S119. doi:10.1016/j.pathol.2018.12.339
- Taylor RW, Pyle A, Griffin H, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. Jama. Jul 2 2014;312(1):68-77. doi:10.1001/jama.2014.7184
- GeneDX. MitoXpanded Panel. GeneDx, Inc. Updated August, 2021. https://providers2.genedx.com/Resources/TIS-Files/TIS-J809.pdf
- GeneDX. Combined Mito Genome Plus Mito Focused Nuclear Gene Panel. https://www.genedx.com/tests/detail/combined-mito-genome-plus-mito-focused-nuclear-gene-panel-736
- ARUP. Mitochondrial Disorders Panel (mtDNA and Nuclear Genes). https://ltd.aruplab.com/Tests/Pub/3001959
- Baylor. Providing insights into unclear health conditions through Mitochondrial testing. Baylor Genetics https://www.baylorgenetics.com/mitochondrial/
- Medical Neurogenetics L. Mitochondrial Genome Sequencing + Deletion Analysis. https://mnglabs.labcorp.com/tests/MOL189/mitochondrial-genome-sequencing-deletion-analysis
- Wood E, Parker M, Dunning M, et al. Clinical long-read sequencing of the human mitochondrial genome for mitochondrial disease diagnostics. 2019;doi:10.1101/597187
- Wagner M, Berutti R, Lorenz-Depiereux B, et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J Inherit Metab Dis. Sep 2019;42(5):909-917. doi:10.1002/jimd.12109
- Legati A, Reyes A, Nasca A, et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochimica et biophysica acta. Aug 2016;1857(8):1326-1335. doi:10.1016/j.bbabio.2016.02.022
- Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: two years' experience with whole-exome sequencing at a national paediatric centre. Journal of translational medicine. Jun 12 2016;14(1):174. doi:10.1186/s12967-016-0930-9
- Kerr M, Hume S, Omar F, et al. MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Molecular Genetics and Metabolism. 2020/09/18/ 2020;doi:10.1016/j.ymgme.2020.08.009
- Kohda M, Tokuzawa Y, Kishita Y, et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS genetics. Jan 2016;12(1):e1005679. doi:10.1371/journal.pgen.1005679
- Fang F, Liu Z, Fang H, et al. The clinical and genetic characteristics in children with mitochondrial disease in China. Science China Life sciences. Jul 2017;60(7):746-757. doi:10.1007/s11427-017-9080-y
- Spath K, Babariya D, Konstantinidis M, et al. Clinical application of sequencing-based methods for parallel preimplantation genetic testing for mitochondrial DNA disease and aneuploidy. Fertil Steril. Jun 2021;115(6):1521-1532. doi:10.1016/j.fertnstert.2021.01.026
- Wu Y, Balasubramaniam S, Rius R, Thorburn DR, Christodoulou J, Goranitis I. Genomic sequencing for the diagnosis of childhood mitochondrial disorders: a health economic evaluation. Eur J Hum Genet. Jun 8 2021;doi:10.1038/s41431-021-00916-8
- Farahvash A, Kassardjian CD, Micieli JA. Mitochondrial Neurogastrointestinal Encephalopathy Disease: A Rare Disease Diagnosed in Siblings with Double Vision. Case Rep Ophthalmol. Jan-Apr 2021;12(1):174-181. doi:10.1159/000514098
- Wen H, Deng H, Li B, et al. Mitochondrial diseases: from molecular mechanisms to therapeutic advances. Signal Transduction and Targeted Therapy. 2025/01/10 2025;10(1):9. doi:10.1038/s41392-024-02044-3
- Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genetics in medicine : official journal of the American College of Medical Genetics. Sep 2015;17(9):689-701. doi:10.1038/gim.2014.177
- Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genetics in medicine : official journal of the American College of Medical Genetics. Dec 2017;19(12)doi:10.1038/gim.2017.107
- Mavraki E, Labrum R, Sergeant K, et al. Best Practice Guidelines for the Molecular Diagnosis of Mitochondrial Disease. Association for Clinical Genomic Science. https://www.acgs.uk.com/media/11935/bpg-for-the-molecular-diagnosis-of-mitochondrial-disease_ratified-november-2020.pdf
- Mancuso M, Arnold M, Bersano A, et al. Monogenic cerebral small-vessel diseases: diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur J Neurol. Jun 2020;27(6):909-927. doi:10.1111/ene.14183
- Hirano M, Carelli V, De Giorgio R, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J Inherit Metab Dis. Aug 9 2020;doi:10.1002/jimd.12300
- Mavraki E, Labrum R, Sergeant K, et al. Genetic testing for mitochondrial disease: the United Kingdom best practice guidelines. European Journal of Human Genetics. 2023;31(2):148-163. doi:10.1038/s41431-022-01249-w
Coding Section
| Code Number |
Code Description |
| 81401 |
Molecular pathology procedure, Level 2 |
| 81403 |
Molecular pathology procedure, Level 4 |
| 81404 | Molecular pathology procedure, Level 5 (e.g., analysis of 2 – 5 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 6 – 10 exons, or characterization of a dynamic mutation disorder/triplet repeat by Southern blot analysis) |
| 81405 | Molecular pathology procedure, Level 6 (e.g., analysis of 6 – 10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11 – 25 exons, regionally targeted cytogenomic array analysis) |
| 81406 | Molecular pathology procedure, Level 7 (e.g., analysis of 11 – 25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26 – 50 exons, cytogenomic array analysis for neoplasia) |
| 81440 |
Nuclear encoded mitochondrial genes (e.g., neurologic or myopathic phenotypes), genomic sequence panel, must include analysis of at least 100 genes, including BCS1L, C10orf2, COQ2, COX10, DGUOK, MPV17, OPA1, PDSS2, POLG, POLG2, RRM2B, SCO1, SCO2, SLC25A4, SUCLA2, SUCLG1, TAZ, TK2, and TYMP |
| 81460 |
Whole mitochondrial genome (e.g., Leigh syndrome, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes [MELAS], myoclonic epilepsy with ragged-red fibers [MERFF], neuropathy, ataxia, and retinitis pigmentosa [NARP], Leber hereditary optic neuropathy [LHON]), genomic sequence, must include sequence analysis of entire mitochondrial genome with heteroplasmy detection. |
| 81465 |
Whole mitochondrial genome large deletion analysis panel (e.g., Kearns-Sayre syndrome, chronic progressive external ophthalmoplegia), including heteroplasmy detection, if performed |
| 81479 | Unlisted molecular pathology procedure |
| 0212U |
Rare diseases (constitutional/heritable disorders), whole genome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, proband |
| 0213U |
Rare diseases (constitutional/heritable disorders), whole genome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, each comparator genome (e.g., parent, sibling) |
| 0214U |
Rare diseases (constitutional/heritable disorders), whole exome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, proband |
| 0215U |
Rare diseases (constitutional/heritable disorders), whole exome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, each comparator exome (e.g., parent, sibling) |
| 0417U | Rare diseases (constitutional/heritable disorders), whole mitochondrial genome sequence with heteroplasmy detection and deletion analysis, nuclear-encoded mitochondrial gene analysis of 335 nuclear genes, including sequence changes, deletions, insertions, and copy number variants analysis, blood or saliva, identification and categorization of mitochondrial disorder-associated genetic variants Proprietary test: Genomic Unity® Comprehensive Mitochondrial Disorders Analysis Lab/Manufacturer: Variantyx Inc. |
| 0532U | Rare diseases (constitutional disease/hereditary disorders), rapid whole genome and mitochondrial DNA sequencing for singlenucleotide variants, insertions/deletions, copy number variations, peripheral blood, buffy coat, saliva, buccal or tissue sample, results reported as positive or negative. |
| 0567U | ORare diseases (Constitutional/heritable disorders), whole-genome sequency analysis combination of short and long reads, for single-nucleotide variants, insertions/deletions and characterized intronic variants, copy-number variants, duplications/deletions, mobile element insertions, runs of homozygosity, aneuploidy, and inversions, mitochondrial DNA sequence and deletions, short tandem repeat genes, methylation status of selected regions, blood, saliva, amniocentesis, chorionic villus sample or tissue, identification and categorization of genetic variants |
| 0657U (effective on 07/01/2026) | Rare diseases (constitutional/heritable disorders), rapid whole genome sequence analysis of comparator nuclear and mitochondrial DNA by next[1]generation sequencing (NGS), using blood or buccal sample, relevant variants reported with proband results Effective on 07/01/2026 |
| 0658U (effective on 07/01/2026) | Rare diseases (constitutional/heritable disorders), rapid whole genome sequence analysis of nuclear and mitochondrial DNA by next-generation sequencing (NGS) for single[1]nucleotide variants (SNVs), insertions/deletions, copy number variants, uniparental disomy, and repeat expansions, using blood or buccal sample, identification and categorization of genetic variants Effective on 07/01/2026 |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 06/08/2026 | Updating Coding Section. Added CPT Codes 0657U and 0658U to be effective 07/01/2026. No other changes. |
| 05/11/2026 | Annual review, no change to policy intent. Updating rationale and references. Updated CPT 0567U verbiage. |
| 02/10/2026 | Annual review, no change to policy intent. Updating policy for clairty and consistency, rationale, and references. |
| 06/12/2025 | Added CPT code 0567U effective 07/01/2025 |
| 02/26/2025 | Adding code 0532U effective 04/01/2025 |
| 01/23/2025 | Annual review, adding new criteria #3 and updating Current criteria #$ to remove exome testing. Also updating description, table of terminology, rationale , references, and coding revision. |
| 01/18/2024 | Annual review, no change to policy intent. Updating description, rationale and references. |
| 09/11/2023 | Updating Coding section. Adding CPT code 0417U effective 10/01/2023. No other changes made. |
| 01/24/2023 | Annual review, no change to policy intent. Policy verbiage updated for clarity. Updating description, rationale and references. |
| 01/12/2022 |
Annual review, no change to policy intent. Updating policy verbiage to clarify acronyms. Also updating rationale and references. |
| 01/22/2021 |
Annual review, reformatting for clarity, updating policy number. Adding medical necessity criteria for mtDNA testing, adding criteria 5 and 6 related to WES testing. Also updating description, rationale, references and coding. |
| 01/02/2020 |
Annual review, no change to policy intent. |
| 09/24/2019 |
Updated coding. No other changes made. |
| 01/10/2019 |
Annual review, updating medical necessity criteria for clarity. |
| 01/25/2018 |
Annual review. Updating medical necessity for clarity of disorders to be tested. No change to policy intent. Updating rationale, references and guidelines. |
| 04/27/2017 |
Annual review, updating category to laboratory. Updating policy verbiage and guidelines related to specific mutations. |
| 04/26/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
| 07/12/2016 |
Annual review, no change to policy intent. Updating background, description, rationale, references and appendix 1. |
| 07/27/2015 |
Annual review, no change to policy intent. Reorganizing policy and guideline verbiage, but NOT changing intent. Updated background, description, rationale and references. Added coding and appendix 1. |
| 07/07/2014 |
New Policy |