
Molecular Testing for Cutaneous Melanoma - CAM 255
Description
Cutaneous melanoma is a common and serious form of skin cancer. Diagnosis of this type of cancer often involves visual examination of the skin by a dermatologist; however, due to the various presentations of nodules, it can be difficult to properly recognize a melanoma case. Although several visual systems have been developed to assist in diagnosis (such as the ABCDE system for signature features), a biopsy is often performed to diagnose a case.1
Genetic testing (particularly gene expression panels) has been explored by researchers to assist in diagnosing cases without a biopsy.2 Another application of genetic testing is “targeted testing”; certain genetic mutations show better patient response with targeted treatment, and it is therefore clinically useful to identify these mutations to help guide selection of the most appropriate therapy.
REGULATORY STATUS
Many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare and Medicaid (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). LDTs are not approved or cleared by the U. S. Food and Drug Administration; however, FDA clearance or approval is not currently required for clinical use.
The FDA-approvals for all the BRAF-targeted therapies include the requirement that BRAF mutation testing be performed by an FDA-approved test.
On August 17, 2011, the U.S. Food and Drug Administration (FDA) announced the approval of Zelboraf (vemurafenib) for unresectable or metastatic melanoma with oncogenic BRAF mutation (V600E). The Cobas® 4800 BRAF V600 Mutation Test was approved as the companion diagnostic for vemurafenib.129
Dabrafenib was FDA-approved in May 2013 for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation, as detected by an FDA-approved test. Dabrafenib is specifically not indicated for the treatment of patients with wild-type BRAF melanoma (Tafinlar (dabrafenib), Jan 2014).
Trametinib was FDA-approved in May 2013 for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test. Trametinib is specifically not indicated for the treatment of patients previously treated with BRAF inhibitor therapy (GlaxoSmithKline. Mekinist Aug 2014).
The companion diagnostic test coapproved for both dabrafenib and trametinib is the THxID™ BRAF Kit manufactured by bioMérieux. The kit is intended “as an aid in selecting melanoma patients whose tumors carry the BRAF V600E mutation for treatment with dabrafenib and as an aid in selecting melanoma patients whose tumors carry the BRAF V600E or V600K mutation for treatment with trametinib” (Genentech, Inc. Zelboraf® March 2014).
The FDA approved the use of the Oncomine Dx target test NGS panel for somatic or germline variants, which includes the BRAF V600E mutation for consideration with dabrafenib therapy as one of the gene variants (Life Technologies Corporation, approved in June 2017).
The FDA approved the FoundationOne CDx NGS panel in November 2017, which does include both the V600E and V600K mutation for possible dabrafenib or vemurafenib therapy (Foundation Medicine, Inc.).
The FDA approved the use of Therascreen BRAF V600E RGQ PCR Kit in April 2020, a real time PCR test that detects BRAF V600E mutations. The Therascreen BRAF V600E RGQ PCR Kit is for use on the Rotor-Gene Q MDx (US) instrument.
New therapies for melanoma continue to receive FDA approval, and the National Cancer Institute summarizes them here: https://www.cancer.gov/about-cancer/treatment/drugs/melanoma.130
Related Policies
CAM 255 Molecular Testing for Cutaneous Melanomas
CAM 288 Testing for Targeted Therapy of Non-Small-Cell Lung Cancer
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- For individuals who have received genetic counseling and who have a diagnosis of cutaneous melanoma, multigene panel testing for cutaneous melanoma-associated likely pathogenic or pathogenic variants (see Note 1, Note 2) is considered MEDICALLY NECESSARY when one of the following conditions is met:
- When the individual has three or more invasive cutaneous melanomas.
- When the individual has invasive cutaneous melanoma and one of the following:
- A personal history of astrocytoma.
- A personal or family history (first-degree relative; see Note 3) with pancreatic cancer.
- For individuals without a diagnosis of melanoma who have received genetic counseling and who are in a family with a known deleterious familial likely pathogenic or pathogenic variant in a cutaneous melanoma-associated gene, the following genetic testing for inherited forms of melanoma is considered MEDICALLY NECESSARY:
- Testing restricted to the known familial likely pathogenic or pathogenic variant.
- Comprehensive genetic testing, including muti-gene panel testing (see Note 1, Note 2), when the specific familial likely pathogenic or pathogenic variant is unknown.
- For individuals without a diagnosis of melanoma who have received genetic counseling, multi-gene panel testing for cutaneous melanoma-associated likely pathogenic or pathogenic variants (see Note 1, Note 2) is considered MEDICALLY NECESSARY only if a family likely pathogenic or pathogenic variant is unknown and the individual has a family history of one of the following:
- Three or more invasive cutaneous melanomas.
- Renal cancer.
- Astrocytoma.
- Uveal melanoma.
- Mesothelioma.
- For individuals who are being considered for molecular-targeted therapy and who have stage III or stage IV cutaneous melanoma, testing of tumor tissue for BRAF, KIT, and NRAS gene mutations (single gene or multi-gene panel testing) is considered MEDICALLY NECESSARY.
- For individuals with a suspicious primary melanocytic skin lesion, risk assessment of the lesion using the DermTech Pigmented Lesion Assay (PLA) (see Note 4) is considered MEDICALLY NECESSARY to help inform a biopsy decision when all of the following conditions are met:
- When lesion size is 5-19 mm;
- When the lesion meets one or more ABCDE criteria (Asymmetry, Border, Color, Diameter, Evolving);
- When the lesion is located where the skin is intact (i.e., non-ulcerated or non-bleeding lesions) and free of psoriasis, eczema, or similar skin conditions;
- When the lesion does not contain a scar;
- When the lesion has not been previously biopsied;
- When the lesion is not already clinically diagnosed as benign or melanoma;
- When the lesion is NOT located on the palms of hands, soles of feet, nails, mucous membranes, or hair covered areas that cannot be trimmed.
- For individuals with a primary cutaneous melanocytic lesion that has not been re-excised, gene expression profiling (GEP) of the lesion with the myPath® Melanoma Assay is considered MEDICALLY NECESSARY when all of the following conditions are met:
- When the lesion has not already been definitively diagnosed as benign or malignant;
- When diagnostic GEP has not already been performed on the same lesion.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For all other situations not described above, genetic testing for inherited forms of cutaneous melanoma is considered NOT MEDICALLY NECESSARY.
- For all other situations not described above, genetic expression profiling tests for cutaneous melanoma is considered NOT MEDICALLY NECESSARY.
- For all situations not described above (e.g., individuals not being considered for targeted therapy, other forms or stages of melanoma), testing for BRAF, KIT, NRAS, and other mutations is considered NOT MEDICALLY NECESSARY.
- Prognostic biomarker testing of a tumor biopsy to assess the risk of melanoma progression (e.g., AMBLor®) is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1: When germline multi-gene panel testing is performed, the panel should at minimum include the following melanoma susceptibility genes: BAP1, CDK4, CDKN2A, MC1R, MITF, PTEN, and TERT.
Note 2: For two or more gene tests being run on the same platform, please refer to CAM 235 Reimbursement Policy.
Note 3: First-degree relatives include parents, full siblings, and children of the individual.
Note 4: While this is not considered a surgical procedure, it is recommended that individuals who are receiving this test should first undergo an informed consent process to discuss the benefits and risks of pursuing this test versus receiving a biopsy to rule out melanoma.
Table of Terminology
| Term |
Definition |
| AAD |
The American Academy of Dermatology |
| ABCDE |
Asymmetry, border, color, diameter and evolving |
| ACD |
Adrenocortical dysplasia protein homolog |
| ACMG |
American College of Medical Genetics and Genomics |
| AMBRA1 |
Autophagy and beclin 1 regulator 1 |
| AMLo |
Loricrin |
| anti-CTLA-4 |
Anti-cytotoxic T-lymphocyte-associated protein 4 |
| ARAF |
Serine/threonine-protein kinase A-Raf |
| ASCO |
American Society of Clinical Oncology |
| ATM |
Ataxia-telangiectasia mutated |
| BAP1 |
Breast cancer susceptibility gene-associated protein-1 |
| BRAF |
B-RAF Proto-Oncogene, serine/threonine kinase |
| BRCA1 |
Breast cancer susceptibility gene |
| BRCA2 |
Breast cancer susceptibility gene 2 |
| CAP |
College of American Pathologists |
| CDK4 |
Cyclin-dependent kinase 4 |
| CDKN2A |
Cyclin-dependent kinase inhibitor 2a |
| CDx |
Companion diagnostic |
| CGH |
Comparative genomic hybridization |
| CHEK2 |
Checkpoint kinase 2 |
| C-KIT |
Proto-oncogene c-KIT |
| CLIA’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CLND |
Completion lymph node dissection |
| CM |
Cutaneous melanoma |
| CMS |
Centers for Medicare & Medicaid Services |
| COVID-19 |
Coronavirus disease 2019 |
| CP-GEP |
Clinicopathological and Gene Expression Profile |
| CRAF |
RAF proto-oncogene serine/threonine-protein kinase |
| ddPCR |
Droplet digital polymerase chain reaction |
| DFS |
Disease-free survival |
| DMFS |
Distant metastasis-free survival |
| DNA |
Deoxyribonucleic acid |
| E |
Glutamate |
| EADO |
European Association of DermatoOncology |
| EDF |
European Dermatology Forum |
| EORTC |
European Organization of Research and Treatment of Cancer |
| ESMO |
European Society for Medical Oncology |
| FAMMM |
Familial atypical multiple mole-melanoma |
| FDA |
Food and Drug Administration |
| FFPE |
Formalin‐fixed, paraffin‐embedded |
| FISH |
Fluorescence in situ hybridization |
| GEP |
Gene expression profile |
| GIST |
Gastrointestinal stromal tumor |
| GWAS | Genome-wide association studies |
| HR |
Hazard ratio |
| KIT |
KIT proto-oncogene, receptor tyrosine kinase |
| LDTs |
Laboratory developed tests |
| LINC00518 |
Long intergenic non-protein coding ribonucleic acid 518 |
| MAS |
Melanoma-astrocytoma syndrome |
| MC1R |
Melanocortin-1 receptor |
| MDM2 |
Mouse double minute 2 |
| MEK |
Mitogen-activated protein kinase/extracellular signal-regulated kinase-1 kinase |
| MITF |
Microphthalmia-associated transcription factor |
| MPWG |
Melanoma Prevention Working Group |
| NBN |
Nibrin |
| NCCN |
National Comprehensive Cancer Network |
| NCI |
National Cancer Institute |
| NED |
Without evidence of disease |
| NER |
Nucleotide excision repair |
| NGS |
Next-generation sequencing |
| NP |
Nurse practitioners |
| NPV |
Negative predictive value |
| NRAS |
Neuroblastoma RAS viral [v-ras] oncogene homolog |
| NSGC |
National Society of Genetic Counselors |
| OS |
Overall survival |
| p14ARF |
Adenosine pyrophosphate -ribosylation factor tumor suppressor |
| p16 |
Cyclin-dependent kinase inhibitor 2A |
| p53 |
Tumor protein P53 |
| PA |
Physician assistants |
| PCR |
Polymerase chain reaction |
| PFS |
Progression-free survival |
| PIK3CA |
Phosphoinositide 3-kinases catalytic subunit alpha |
| PLA |
Pigmented lesion assay |
| PLA(−) |
Pigmented legion assay negative |
| POT1 |
Protection of telomeres 1 |
| PRAME |
Preferentially expressed antigen in melanoma |
| PTEN |
Phosphatase and tensin homolog |
| qPCR |
Quantitative polymerase chain reaction |
| RB1 |
Retinoblastoma protein |
| RCM |
Reflectance confocal microscopy |
| RFS |
Recurrence-free survival |
| RNA |
Ribonucleic acid |
| ROC |
Receiver operating characteristic |
| RTK |
Receptor tyrosine kinase |
| RT-PCR |
Real-time polymerase chain reaction |
| SIC |
Satisfactory disease free |
| SIGN |
Scottish Intercollegiate Guidelines |
| SLN |
Sentinel lymph node |
| SLNB |
Sentinel lymph node biopsy |
| SLNBx |
Sentinel lymph node biopsy |
| SSE |
Skin self-examination |
| SSO |
Society of Surgical Oncology |
| TERT |
Telomerase reverse transcriptase |
| TMB |
Tumor mutation burden |
| TN |
True-negative |
| TP |
True-positive |
| UV |
Ultraviolet |
| WRN |
Werner's syndrome protein |
| WT |
Wild type |
| XP |
Xeroderma pigmentosum |
| XPC |
Xeroderma pigmentosum group c |
| XPD |
Xeroderma pigmentosum group d |
Rationale
Skin cancer is the most common form of cancer, arising from the metaplastic transformation from any of the cell types of the skin.3 Melanomas, which develop from the pigment-producing melanocytes, although much less prevalent than non-melanoma skin cancer, are increasing in incidence.4,5 Early and accurate diagnosis is essential, as late-stage melanoma is among the most fatal forms of skin cancer.6 This, however, presents a significant challenge due to the difficulty of interpreting the histopathology of melanoma and the resulting interobserver and intra-observer variability.7,8
Genetic Testing for Familial Cutaneous Melanoma
A family history of melanoma is reported by about 10% of melanoma patients,9 and inherited germline mutations reportedly “increase melanoma risk from 4- to >1000-fold.”10 Determining the genetic origin, however, is complicated, as a portion of familial melanoma can be attributed to shared sun exposure experiences in family members with susceptible skin types.11
The majority of familial cases lack identifiable germ-line mutations in either known susceptibility genes or in genes commonly mutated in sporadic melanoma.12 Uncommon, but high-risk, alleles have been found to contribute to the hereditary cancer phenotype that includes unilateral lineage, multi-generational, multiple primary lesions, and early onset of disease.9 Additional research has identified a relationship between telomere length and familial melanoma; patients with familial melanoma had longer telomeres compared to patients with sporadic melanoma.13 As such, genes such as TERT—which encodes for the catalytic subunit of telomerase—and proteins such as POT1—a shelterin complex protein—have been implicated in the presence of multiple primary melanomas and early-onset melanoma in a subset of high-density families.14 Notably, a novel POT1 mutation was reported in a family with multiple primary melanomas and other cancer types, suggesting that POT1 mutations may contribute to a broader hereditary cancer predisposition beyond melanoma.15
Cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase 4 (CDK4) are the most identified gene mutations in familial forms of melanoma, which can be defined as a family where either two first-degree relatives or three or more melanoma patients on the same side of the family are diagnosed with melanoma. Germline CDKN2A mutations have been identified in approximately 20-40% of familial melanoma cases where three or more family members are affected.14,16 CDKN2A encodes several proteins involved in cell cycle regulation, including p16, which inhibits CDK4,17,18 and p14ARF, which inhibits MDM2 from regulating p53.19 The CDKN2A gene is located on chromosome 9p21.3 and, through the use of two first exons (1α and 1β), encodes the p16^INK4A^ (156 amino acids) and p14^ARF^ interacts with the retinoblastoma protein to control the G1-S transition of the cell cycle, while p14^ARF^ induces cell cycle arrest in G2 and promotes apoptosis. These proteins are often simultaneously affected in malignant melanoma, which explains the loss of multiple checkpoint controls in tumor progression.15
Germline CDKN2A mutations in melanoma families are usually missense or nonsense changes that impair the function of the p16 protein, allowing for unchecked cell cycle progression; however, rare mutations in the p14ARF protein have also been reported and result in proteasomal degradation of p53 with subsequent accumulation of DNA damage.20 Overall survival is worse in those with a germline CDKN2A mutation than those with sporadic melanoma or familial melanoma with wild-type CDKN2A genes. Germline mutations also predisposed individuals to an increased number of malignancies, such as pancreatic and lung cancer.21
Mutations in CDKN2A/p16 are associated with familial atypical multiple mole-melanoma (FAMMM syndrome), which is characterized by numerous nevi (some atypical), a family history of melanoma, and an increased risk of pancreatic cancer.22,23 Carriers of a FAMMM mutation typically present with cancer at a younger age than non-carriers.24 Mutations in p14ARF are linked to Melanoma-Astrocytoma Syndrome (MAS), a variant of FAMMM characterized by both cutaneous melanomas and nervous system tumors.25 Inheritance of CDKN2A mutations are autosomal dominant, but these mutations have variable penetrance based on sun exposure patterns and coinheritance of other melanoma-associated variants, conferring a 76% lifetime risk of developing melanoma in the US.26,27 Mutations in CDK4 are even less common, but were most often found affecting arginine 24, resulting in a CDK4 protein that is insensitive to inhibition by the p16 protein. No apparent differences exist in the phenotype (e.g., age at diagnosis, number of melanomas) of families carrying either CDKN2A or CDK4 mutations. In aggregate, between 20-45% of familial melanomas are associated with germline mutations in CDKN2A or CDK4.22,28
Other rare mutations have been associated with melanoma. Germline variations in the melanocortin-1 receptor (MC1R) gene alter the risk of melanoma in individuals with and without CDKN2A mutations.20,29,30 Genome-wide association studies (GWAS) have identified multiple SNPs in pigmentation-related genes, including ASIP, TPCN2, IRF4, OCA2, and TYRP1, that interact with environmental UV exposure to modify melanoma risk. For instance, ASIP shifts melanin production to a less productive isoform, and TPCN2 polymorphisms are associated with increased melanoma risk, particularly in males.15 Germline variants in genes that encode for BRCA1-associated protein-1 (BAP1), telomerase reverse transcriptase (TERT), and microphthalmia-associated transcription factor (MITF) have also been added to the list of genes harboring familial melanoma-predisposing mutations.31,32 These are more often associated with “mixed cancer syndrome,” where melanoma may appear in the context of a more general predisposition for malignancy. The BAP1 tumor syndrome is associated with the appearance of cutaneous melanoma, uveal melanoma, and various internal malignancies.33 Mutations in the promoter region of TERT, the protein component of telomerase, and in various components of the shelterin complex have been associated with a higher incidence of melanoma and other internal malignancies.34,35 Mutations in MITF are associated with a higher nevus count, cutaneous malignant melanoma onset before 40 years of age, and non-blue eye color with no association to freckling, skin color, or hair color.36,37 Xeroderma pigmentosum (XP) is a rare disorder in which patients have a mutation in genes involved in nucleotide excision repair (NER). Patients with mutations in XPC and XPD have an increased risk of melanoma.38 Lastly, Cowden syndrome, a type of PTEN hamartoma tumor syndrome characterized by the appearance of trichilemmomas, papillomatous papules, mucosal lesions (papules) and palmar-plantar keratosis within the first three decades of life, is associated with a higher risk of melanoma.31,39
Additional susceptibility genes have been reported, such as MGMT, which encodes a DNA repair enzyme and has been associated with melanoma predisposition as well as promoter methylation in melanoma brain metastases.15
Several proprietary gene panels exist for assessment of familial cutaneous melanoma. For example, the DermTech Pigmented Legion Assay (PLA) leverages their “Smart Sticker” technology to remove cellular material from the stratum corneum, the material of which is then analyzed for over-expression of LINC00518 (long intergenic non-coding RNA 518) and PRAME (preferentially expressed antigen in melanoma), which may be incidental with genomic atypia associated with melanomas.40 Moreover, Invitae offers a 12-gene panel (nine primary genes plus three genes with “preliminary evidence for melanoma”), Fulgent offers a 14-gene panel, and GeneDx offers a 9-gene panel.41-43 These panels may include genes traditionally associated with familial melanoma itself (such as CDKN2A) as well as genes whose variants are not primarily associated with familial melanoma, but confer added risk regardless.21
Clinical Utility and Validity of Genetic Testing for Familial Cutaneous Melanoma
The frequency of CDKN2A mutations in patients with a single primary melanoma or multiple primary melanoma were 1.2% and 2.9%, respectively;44 however, depending on selection criteria, mutation frequency rates of CDKN2A can range from 5% to 72%45 with a family history of melanoma considered the most important risk factor. The established rule of three is used when proposing genetic testing for primary melanomas; it is generally understood that when three or more melanomas or genetically related cancers are identified in the same patient, or in first- and second-degree relatives, the pretest probability is increased above ten percent and the cost of genetic screening can be justified.46
In a study on CDK2NA genetic testing conducted in Sweden between 2015 and 2020, 913 members across 403 families were identified with cutaneous melanoma. Pissa, et al. (2021) found that melanoma cases found in families testing positive for pathogenic variants of CDK2NA boasted significantly higher mortality rates, such that families with the variants had 37.6% melanoma cases that died from melanoma as compared to the 10.0% without (p<0.001), independent of age, sex, and tumor stage. This significant melanoma-specific mortality associated with families with CDK2NA variants is motivation to identify and enroll carrier families in a preventive surveillance program.47 However, a potential confounding variable involves the diagnoses of pancreatic cancers in the subjects in the study; as pathogenic strains of CDK2NA are also associated with cancers other than melanomas, the 129 of the 913 members present a limitation to the specificity of the study as there did not appear to be a clear manner of segregating its influence.
Stolarova, et al. (2020) analyzed 264 Czech melanoma patients with early onset, double primary tumors or family history by next generation sequencing NGS analysis of 217 genes, and they identified that “mutations in high-to-moderate melanoma risk genes and in other cancer syndrome genes were significantly associated with melanoma risk,” with those genes including CDKN2A, POT1, and ACD for high-to-moderate melanoma risk, and NBN, BRCA1/2, CHEK2, ATM, WRN, and RB1 for other cancer syndrome genes. An increased potential of carrying mutations was found in “patients with double primary melanoma, melanoma and other primary cancer, but not in patients with early age at onset.”48 CDK2NA was the most frequently mutated gene among those with high-to-moderate risk, and in other studies reviewed by Stolarova, et al. (2020), there was an increased risk for pancreatic cancer among families with CDK2NA mutation, and a more established family history.
Leachman, et al. (2017) published an updated algorithm for the identification, testing, and management of hereditary melanoma; the rule of three has been incorporated into this algorithm as an indication for genetic testing in multiple melanomas. The researchers state that “Any patient or family that meets the updated rule of threes should be considered a candidate for genetic testing. If melanoma is the only cancer in a pedigree, then to meet the threshold of genetic testing, a pedigree should have three primary melanomas in first- or second-degree relatives in areas with a high melanoma incidence or two primary melanomas in a low-incidence area. This melanoma panel should include BAP1, CDK4, and CDKN2A. Genes for which risk has not been established but for which studies suggest an elevated risk include MITF and POT1 and we recommend including these in the melanoma panel.”46
Genetic testing for commonly known cutaneous melanoma mutations can be utilized to determine prognosis and overall survival. Aoude, et al. (2020) found that “germline mutation status was the most significant biomarker for OS [overall survival]” and “survival outcomes for germline carriers are poor with the current standards of care.” When using BRAF status and tumor mutation burden (TMB) for prognosis of cutaneous melanoma patients, “BRAF V600 wild-type patients had significantly longer PFS [progression-free survival] than the V600 mutant group (p = 0.0317) … For stage III/IV resected patients, TMB was also significantly associated with longer PFS (p = 0.0034).”49 The greater the number of recognizable mutations, the more targeted attacks against cancerous cells can be made and the better the prognosis.
Genetic Testing for Acquired Cutaneous Melanoma
Melanoma is a particularly lethal and aggressive cancer with the ability to metastasize to any organ.50 Cutaneous tumors as little as one mm in thickness are capable of lymph node metastasis (Stage III), resulting in a significant decrease in the five-year survival rate from 90% to 56%.51 If the cancer has spread beyond the lymph nodes (Stage IV), an even more dramatic decrease in five-year survival to 15% will occur.52 Genetic testing for acquired melanoma can identify the activating genetic alterations.
Genetic testing is important to determine the most efficient treatment method for a melanoma patient. Despite the high frequency in nevi, the role of BRAF mutations in oncogenesis is well established53 and has been confirmed in clinical trials.54 BRAF is a member of the RAF family of protein serine/threonine kinases (ARAF, BRAF, CRAF) that is activated by Ras proteins during intracellular signaling cascades. Mutations in BRAF appear to be the most common genetic alteration in melanoma55 and occur more frequently in melanoma than lung, colon, and ovarian carcinoma.52 The remarkable efficacy of BRAF inhibitors led to the accelerated approval of vemurafenib for unresectable and metastatic melanoma. Importantly, BRAF mutation testing is warranted for determining therapeutic eligibility as selective BRAF inhibitors pose significant risk of cutaneous squamous cell carcinoma and have the potential to increase disease progression in BRAF wild type (mutation negative) tumors.52
The mutations, BRAF, KIT, PIK3CA and NRAS, are commonly seen in melanoma cases56,57 with an estimated 50% of melanomas exhibiting the BRAF V600E mutation.58
Table 1. Frequency of Melanoma Subtypes with Activating Genetic Alterations in BRAF and KIT (taken from Grossmann, et al. (2012))
| Aberration |
Cutaneous −CSD |
Cutaneous + CSD |
ALM |
MM |
| BRAF mutation |
53% |
8% – 11% |
10% |
15% |
| KIT mutation; amplification |
0 |
17% |
11% – 38%; 19% – 27% |
6% – 19%; 20% – 33% |
A variety of methods are utilized for BRAF and KIT mutational analysis testing in melanoma, which has resulted in no standardized procedures for testing. Because numerous techniques are available and updated methods continue to be released, labs have been reluctant to switch BRAF platforms to accommodate one specific drug for one disease.52 Current BRAF genetic testing methods include BRAF V600E by real-time PCR, BRAF V600E mutation only by Sanger sequencing, BRAF full gene sequence analysis, and BRAF next generation sequencing.59
However, a recent study found good overall compliance of labs with the College of American Pathologists (CAP)60 and National Comprehensive Cancer Network (NCCN) guidelines for molecular diagnosis of tumors61 despite not using the specific FDA-approved test.
Table 2. Molecular Testing Adherence to NCCN Guidelines (taken from Volmar, et al. (2015))
|
|
All Institutions Percentiles |
|||||
| n |
10th |
25th |
Median |
75th |
90th |
|
| Retrospective study (lung, colorectal, melanoma) |
||||||
| Percentage of tests that strictly meet the guideline |
26 |
32.6 |
64.7 |
70.9 |
82.7 |
89.7 |
| Percentage of tests that at least loosely meet the guideline |
26 |
57.4 |
90.7 |
95.1 |
98.9 |
100.0 |
| Prospective study (all case type) |
||||||
| Percentage of tests that strictly meet the guideline |
23 |
20.0 |
31.4 |
53.3 |
66.7 |
70.5 |
| Percentage of tests that at least loosely meet the guideline |
23 |
75.0 |
87.0 |
94.3 |
100.0 |
100.0 |
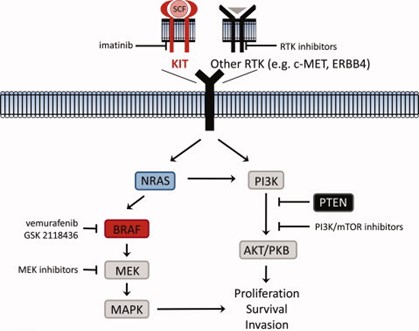
Historically, systemic therapy for metastatic melanoma provided very low response rates and little to no benefit in overall survival.62,63 However, this is beginning to change. For example, researchers have now identified that those with the BRAF V600 mutation are able to obtain both immune checkpoint inhibitor therapy and other targeted therapies for an improved overall treatment regimen.64 Moreover, the immune-boosting anti-CTLA-4 antibody ipilimumab65 and testing and development of small molecule kinase (KIT and BRAF) inhibitors have yielded improvements in long-term survival for melanoma patients.53,66,67
Figure 1: Imatinib and RTK Inhibitor Pathways for Melanoma Treatment (image taken from Grossmann et al., 2012)

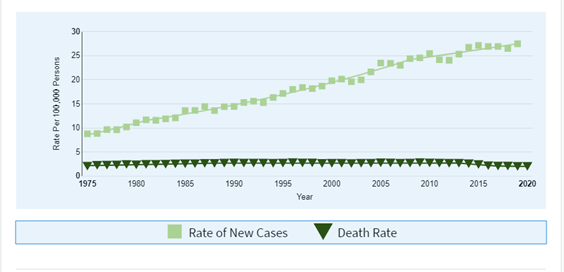
The lifetime risk for the general population of developing melanoma is one in 5568 and that risk has increased approximately two percent annually since 1960.69 National Cancer Institute now reports that melanoma of the skin represents 5.2% of all new cancer cases in the United States; approximately 99,780 Americans in total will be diagnosed with melanoma in 2022.68
In 2022, the number of new cases of melanoma of the skin per 100,000 people was 21.5. Further, the number of melanoma patient deaths in 2022 was 2.1 per 100,000 individuals, accounting for 1.3% of all cancer deaths in United States.68
Figure 2. New Cases and Deaths of Melanoma of the Skin per 100,000 Persons (taken from NCI (2023))
Clinical Utility and Validity of Genetic Testing for Acquired Cutaneous Melonama
O'Brien, et al. (2017) analyzed samples to determine if BRAF mutation identification by immunohistochemistry was a suitable alternative to PCR. The study included 132 patients, and the anti-BRAF V600E VE1 clone antibody was used for immunohistochemistry detection. A sensitivity of 86.1% and specificity of 96.9% was shown with the anti-BRAF V600E VE1 clone antibody; “The concordance rate between PCR and immunohistochemical BRAF status was 95.1% (116/122).”70 As both methods were in high agreement, immunohistochemistry may be a viable alternative to PCR for BRAF mutation testing.
Corean, et al. (2019) utilized several different techniques on metastatic melanoma samples, including bone marrow morphology, histology, immunophenotyping, molecular genetic testing and BRAF V600E immunohistochemistry. BRAF immunohistochemistry was detected in two patients, and molecular testing confirmed these results; researchers then stated that “BRAF V600E immunohistochemistry is useful as a surrogate marker of molecular results,” once again highlighting the fact that immunohistochemistry may be a viable alternative for BRAF mutation testing.71
Available BRAF tests and analytical sensitivities:
- “The BRAF V600E by real-time PCR test uses a TaqMan® Mutation Detection Assay to detect the V600E mutation in exon 15 of BRAF in tumor (somatic) cells. The sensitivity of the TaqMan assay is ~0.1% mutant DNA in a wild-type background. Poor DNA quality, insufficient DNA quantity or the presence of PCR inhibitors can result in uninterpretable or (rarely) inaccurate results.
- The BRAF (V600E) mutation only by Sanger sequencing uses a DNA-based PCR-sequencing assay to detect the V600E in exon 15 of BRAF. The limit of detection for Sanger sequencing is >20% mutant DNA in a wild-type background.
- The BRAF full gene sequence analysis test uses a DNA-based PCR-sequencing assay to detect point mutations in the coding sequence and intron/exon boundaries of the BRAF gene. The sensitivity of DNA sequencing is over 99% for the detection of nucleotide base changes, small deletions and insertions in the regions analyzed. Rare variants at primer binding sites may lead to erroneous results. The limit of detection for Sanger sequencing is >20% mutant DNA in a wild-type background.”59
- The BRAF NGS with TruSeq had sensitivity of over 99%.72
Colombino, et al. (2020) compared BRAF mutation testing performed with conventional nucleotide sequencing approaches (Sanger sequencing and pyrosequencing) with real-time polymerase chain reaction (RT-PCR) or next-generation sequencing (NGS) assays to assess the levels of concordance between these various techniques. There were 319 tissue samples analyzed and initially screened with conventional approaches. The initial screen found pathogenic BRAF mutations in 144 (45.1%) cases. RT-PCR (Idylla™ BRAF mutation assay) detected 11 (16.2%) and three (4.8%) additional BRAF mutations after Sanger sequencing and pyrosequencing, respectively. NGS detected one additional BRAF-mutated case (2.1%) among 48 wild-type cases previously tested with pyrosequencing and RT-PCR. According to the data, RT-PCR is more accurate than both Sanger sequencing and pyrosequencing in detecting BRAF mutations. Overall, RT-PCR had a good concordance with the other tests; "real-time PCR is a rapid method which achieves the same maximum level of sensitivity of NGS (up to 98%), without requiring particular skills.”73 According to the author, “[RT-PCR and NGS] improved the diagnostic accuracy of BRAF testing via the detection of additional BRAF mutations in a subset of false-negative cases previously tested with Sanger sequencing or pyrosequencing. In attendance of further confirmations in larger prospectively designed studies, the use of two sensitive molecular methods may ensure the highest level of diagnostic accuracy.”73
The BRAF analysis is an accepted medical practice for patients with unresectable, metastatic stage IV melanoma.74 However, recent randomized controlled trials have indicated the benefits of expanding BRAF analysis. A study published in The Lancet Oncology by Amaria, et al. (2018) compared standard of care in patients with high-risk, surgically resectable melanoma (stage III or IV) to similar patients receiving a regimen of a neoadjuvant plus adjuvant dabrafenib and trametinib. All patients had to be of confirmed BRAF V600E or BRAF V600K status to participate in either the control or experimental groups. In the follow-up (median of 18.6 months), 10/14 (or 71%) of patients in the experimental group remained event-free (i.e., alive without disease progression), whereas zero out of seven (0%) of the control group receiving standard of care remained event-free. The authors conclude that the “Neoadjuvant plus adjuvant dabrafenib and trametinib significantly improved event-free survival versus standard of care in patients with high-risk, surgically resectable, clinical stage III-IV melanoma.”75
Another study published by Zippel, et al. (2017) researched the use of perioperative BRAF inhibitors on patients with stage III melanoma. All patients had to be confirmed BRAF V600E to participate in the study. Of the thirteen patients, twelve “patients showed a marked clinical responsiveness to medical treatment, enabling a macroscopically successful resection in all cases”; moreover, “at a median follow up of 20 months, 10 patients remain free of disease.”76 One patient died prior to surgery in this study. The authors conclude that the “Perioperative treatment with BRAF inhibiting agents in BRAFV600E mutated Stage III melanoma patients facilitates surgical resection and affords satisfactory disease free (sic) survival.”76
Gene Expression Profiling and Biomarkers for Diagnosis and Prognosis
Currently, patients presenting with a suspicious pigmented lesion undergo excisional biopsy which is then subjected to histopathologic examination by a pathologist.74 The majority of melanocytic neoplasms can be accurately classified by this approach; however, in some cases confidently differentiating benign melanocytic nevi from malignant melanoma can be extremely difficult or impossible despite additions to histopathologic assessment, such as the evaluation of Breslow depth.4,77 In these cases, even diagnoses from expert pathologists can be discordant 7,8,78 and subject to diagnostic drift.79
A number of diagnostic and prognostic genetic tests for melanoma have been developed as ancillary tests to assist in this differentiation and resultant risk stratification,4 including microRNAs as biomarkers to distinguish between melanomas and nevi.80 Gene expression profiling is growing in popularity for the diagnosis and prognosis of cutaneous melanoma. Molecular tests based on gene expression profiling of cutaneous melanoma are commercially available. They include the myPath Melanoma,81 2-GEP Pigmented Lesion Assay,82 DecisionDx-Melanoma,83 and a clinicopathologic (CP)-GEP model by SkylineDx.84
Another risk stratification test is the melanoma prognostic test, AMBLor®, used to identify low-risk non-ulcerated early-stage cutaneous melanoma in children and adults. The AMBLor® test looks for the presence of two prognostic biomarker proteins, AMBRA1 and loricrin, both of which are found in the skin overlying the tumor. Melanomas at a lower risk of recurrence/spread continue to express AMBRA1 and/or loricin in the tumoral epidermis. In contrast, tumors that are at a higher risk lose the expression of both proteins. 85
Clinical Utility and Validity of Gene Expression Profiling and Biomarkers for Diagnosis and Prognosis
myPath Melanoma (Castle Biosciences)
A 23-gene expression profile and algorithm that assigns various weights and thresholds of expression for each gene was developed to differentiate benign melanocytic nevi from malignant melanoma; this gene expression profile and algorithm was determined to have a sensitivity of 89% and specificity of 93%.86 Further, three experienced dermatopathologists validated this method against an independent histopathologic evaluation; a sensitivity of 91.5% and a specificity of 92.5% was determined.87 This gene expression profile was developed into a commercial test known as myPath Melanoma, which generates a single numerical score along with a classification of likely malignant, likely benign, or indeterminate, for a given cutaneous lesion. Across various validation studies, myPath Melanoma has demonstrated a sensitivity in the range of 90-94% and a specificity in the range of 91-96%.88
myPath Melanoma correlates closely with long-term clinical outcomes by adding valuable adjunctive information to aid in the diagnosis of melanoma. The test is “designed to provide objective information to aid in the diagnosis and to inform management decisions for patients with ambiguous melanocytic lesions.”89 An examination of the utility of this test6 found that the results of this gene expression signature have a significant clinical impact with 71.4% (55/77) of cases changing from pretest recommendations to actual treatment. Majority of changes were consistent with the test result. There was an 80.5% (33/41) reduction in the number of biopsy site re-excisions performed for cases with a benign test result. However, when more challenging samples were included with 39 histopathologically unequivocal lesions (15 malignant, 24 benign) and 78 challenging lesions interpreted by expert consensus (27 favor malignant, 30 favor benign, and 21 ambiguous), myPath Melanoma had a lower sensitivity and specificity than fluorescence in situ hybridization (FISH) (FISH: 69% sensitivity, 91% specificity; myPath; 55% sensitivity, 88% specificity).90
In contrast, Clarke, et al. (2020) completed another study that evaluated the accuracy of myPath melanoma in diagnostically uncertain neoplasms. Out of 125 diagnostically “uncertain” cases, myPath melanoma demonstrated a sensitivity of 90.4%. In this study, the test displayed no significant difference in sensitivity between the diagnostically uncertain (equivocal) and unequivocal lesion groups.
Pigmented Lesion Assay (PLA) (DermTech)
DermTech has developed a pre-diagnostic quantitative polymerase chain reaction (qPCR)-based pigmented lesion assay (PLA) that measures the expression of two genes in the stratum corneum to assist with diagnostic or prognostic information for potential melanoma cases.92 DermTech’s PLA identifies malignant changes on a genomic level that cannot be detected with the human eye; this assay can be used to support clinicians in their decision to biopsy suspicious nevi. This test has the potential to increase the number of early melanomas biopsied and reduce the number of benign lesions biopsied, thereby improving patient outcomes.93 A recent study has given this pigmented lesion assay a sensitivity of 91-96%, a specificity of 69-91%, and a negative predictive value of approximately 99%.94
To help support clinicians in their decision to biopsy, this noninvasive two gene expression assay of the LINC00518 and PRAME genes has been developed for use on adhesive patch biopsies. Skin sampling via an adhesive patch allows for DNA, RNA, skin tissue and microbiome samples to be safely obtained and transported cost effectively by mail at room temperature; skin cells, T-cells, dendritic cells, melanocytes and other types of cells can be analyzed by this method.95 Further, this technique is a much more cost-effective option. Hornberger and Siegel (2018) report that PLA testing could save approximately $447 per lesion compared to traditional biopsies.
Gerami, et al. (2017) tested the validity of the two-gene panel based on LINC00518 and PRAME on differentiating melanoma from nonmelanoma in a multicenter study across 28 sites in the United States, Europe, and Australia. In a sample of 398 (87 melanomas and 311 nonmelanomas), it was found that this classification method was able to accurately identify melanomas from nonmelanomas with a sensitivity of 91% and a specificity of 69%. An application study of 381 patients found that the estimated real-world sensitivity of the DermTech PLA was 95% and specificity was 91%;97 overall, 93% of PLA results positive for both LINC00518 and PRAME were diagnosed histopathologically as melanoma. Further, this study was also used to identify if the real-world clinical use of the DermTech PLA could change physician behavior and reduce the overall number of biopsies performed. The PLA identified 51 PLA(+) test results, and 100% of these pigmented skin lesions were biopsied (37% were melanomas). Furthermore, “Nearly all (99%) of 330 PLA(-) test results were clinically managed with surveillance. None of the three follow-up biopsies performed in the following three to six months, were diagnosed as melanoma histopathologically.”97 The PLA test altered clinical management of pigmented lesions and shows high clinical performance. However, it is uncertain if the negative samples—as determined by the PLA—will remain so, as “we [the authors] cannot rule out that some PLA(−) lesions may not have been adequately reassessed in the follow-up period and we certainly recommend erring on the side of caution and surgically biopsying a lesion in question if additional risk factors, further clinical suspicion, or patient concern mandate such a step.”97
The PLA test has also been validated against driver mutations in melanoma, including BRAF, NRAS, and TERT. Ferris, et al. (2019) studied the mutation frequency of these genes using samples obtained from the PLA technique (e.g., adhesive patch) in both histopathologically confirmed melanomas (n = 30) and non-melanoma controls (n = 73). “The frequency of these hotspot mutations in samples of early melanoma was 77%, which is higher than the 14% found in nonmelanoma samples (P < 0.0001). TERT promoter mutations were the most prevalent mutation type in PLA-positive melanomas; 82% of PLA-negative lesions had no mutations, and 97% of histopathologically confirmed melanomas were PLA and/or mutation positive.”98 A total of 86% of the non-melanomas within this validation cohort contained no mutations. The authors next analyzed 519 real-world PLA samples for the same mutations. Similar to the previous validation cohort, 88% of this larger cohort also contained no mutations, indicating that the PLA test can rule out lesions with few mutational risk factors for melanoma.
This further confirmed the “real-world” negative predictive value (NPV) and positive predictive value (PPV) of the PLA, “by following a cohort of 1,233 PLA-negative pigmented lesions for evidence of malignancy for up to 36 months and by re-testing a separate prospective cohort of 302 PLA-negative lesions up to two years after initial testing.” Out of this total PLA-negative cohort, 1,233 lesions had a confirmed follow-up evaluation; of these, ten melanomas were subsequently detected by way of biopsy, resulting in an NPV of 99.2%. Out of 316 PLA-positive cases, 59 were diagnosed as melanoma by histopathology, resulting in a PPV of 18.7%.99 The PPV achieved through use of the PLA may represent a five-fold improvement over the current standard according to the test’s developers.
In a continuing medical education series, Skudalski, et al. (2022) confirm that the PLA is to be used as a risk stratification aid in determining whether to biopsy a skin lesion. The authors recommend that in the case of a positive PLA result, “biopsy in toto should be performed immediately.” They further comment that after a negative PLA result, clinicians have chosen to follow up within 12 months to further evaluate the skin lesion; however, a biopsy in toto should be considered if a patient remains concerned about a lesion, or if the lesion has evolved.100
A recent report by Robinson and Jansen (2020) outlined a proof-of-concept pilot program of remote physician-guided self-sampling (i.e. telehealth administration of the adhesive patch) during the Illinois stay-at-home order of the COVID-19 pandemic. The authors also surveyed skin self-examination (SSE) anxiety as well. Two cohorts were used in this pilot, an experimental group (n=7), and a randomly selected physician-sampled control case group (n=10). The authors report that SSE-induced anxiety has increased during the COVID-19 pandemic. It should be noted that surveys were administered to much larger groups than those administering self-sampling tests. There were 258 surveys about SSE anxiety conducted prior to the COVID-19 pandemic, and 211 surveys during the COVID-19 pandemic. The authors state, “Guided self-sampling led to molecular risk factor analyses in 7/7 (100%) of cases compared to 9/10 (90%) randomly selected physician-sampled control cases... Adhesive patch self-sampling under remote physician guidance is a viable specimen collection option.”101
DecisionDx-Melanoma (Castle Biosciences)
After a melanoma case has been identified, several management approaches may be considered. However, best melanoma management practices are constantly evolving. A common technique to assess the spread of a tumor, such as melanoma, is a sentinel lymph node biopsy (SLNB). This procedure is used to evaluate whether the cancer has spread beyond the original tumor site and into the lymphatic system. The lymphatic or lymph system transports fluid known as lymph throughout the body; this fluid contains white blood cells which help to fight infections. The lymphatic system also aids in ridding the body of other waste and toxins. The SLNB technique essentially helps the physician to stage the tumor.
However, melanoma-related lymph node spread is very complex and is associated with many factors, including age, location, thickness, ulceration, gender, and regression.102
The DecisionDx-Melanoma test is a gene expression profile (GEP) test that measures the expression of 31 different genes in a tumor tissue sample. This test was designed for cutaneous melanoma patients undergoing or considering SLNB and can help to identify the risk of cancer recurrence or metastasis in stage I-III melanoma.83 DecisionDx-Melanoma may help physicians guide treatment options, including whether to perform a SLNB in eligible patients, and what type of follow up treatment is necessary.83
The DecisionDx-Melanoma is performed on tumor tissue biopsies that have been preserved as formalin‐fixed, paraffin‐embedded (FFPE) samples.83 Researchers have highlighted that the identification of tissue-based prognostic markers in melanoma has been a challenging obstacle for researchers, as “One major limitation is that most primary melanomas are preserved as formalin‐fixed, paraffin‐embedded (FFPE) samples rather than fresh‐frozen tissues because of the small size of the specimens.”103 High-quality genetic material is more challenging to extract from FFPE samples, and messenger RNA is inconsistent in FFPE samples.103
The prognostic utility of this test has been measured by Keller, et al. (2019); a total of 159 patients participated in this study and were followed up with, on average, 44.9 months after initial testing. Gene expression profiling results helped to categorize patients into two groups: low-risk patients were placed in Class 1, and high-risk patients in Class 2; 117 patients were placed in Class 1 and 42 patients in Class 2.104 Results showed that this gene expression profiling test had great prognostic abilities. “Gender, age, Breslow thickness, ulceration, SNB positivity, and AJCC stage were significantly associated with GEP classification (P < 0.05 for all). Recurrence and distant metastasis rates were five percent and one percent for Class 1 patients compared with 55% and 36% for Class 2 patients. Sensitivities of Class 2 and SNB for recurrence were 79% and 34%, respectively.”104
Berman, et al. (2019) gathered an expert panel of nine dermatologists/dermatologic surgeons/ dermatopathologists and completed 29 clinical scenarios in which gene expression tests could be used appropriately; several gene expression profiling (GEP) tests were used including a 2-GEP assay, 23-GEP assay and 31-GEP assay. “The 2-GEP assay for melanoma diagnosis received one B-strength and six C-strength recommendations. The 23-GEP diagnostic test received one A-strength, three B-strength, and four C-strength recommendations. The 31-GEP prognostic assay received one A-strength, seven B-strength, and six C-strength recommendations”; this report shows that the 31-GEP assay received the highest overall recommendations by the expert panel.105
Another study reported that the 31-GEP was validated in almost 1600 patients “as an independent predictor of risk of recurrence, distant metastasis and death in Stage I-III melanoma and can guide SLNB decisions in patient subgroups, as demonstrated in 1421 patients”; further, an appropriate 31-GEP testing population was identified and concluded that it is best used on patients with cutaneous melanoma tumors greater than or equal to 0.3 mm thick.106 However, this study reports several conflicts of interest that are important to note as multiple authors are employees at Castle Biosciences, Inc., and one author is a consultant and speaker for the company.106
Greenhaw, et al. (2020) completed a meta-analysis of the DecisionDx-Melanoma 31-GEP prognostic test in a total of 1,479 patients. This meta-analysis included participants from three different studies. The patient analysis showed that the five-year recurrence and distant metastasis-free survival rate for Class 1A patients was 91.4% and 94.1% respectively and were 43.6% and 55.5% for Class 2B patients. The 31-GEP was then used to estimate the likelihood of recurrence and distance metastasis. The GEP test exhibited a sensitivity of 76% for each endpoint, showing consistency and accuracy for the identification of at increased risk of metastasis.107
Moreover, Marchetti, et al. (2020) completed an assessment of the prognostic accuracy of the 31-GEP in patients that had cancer labeled as AJCC state I or II disease. Patients with localized melanoma were included from external validation studies, and after exclusion criteria, there were seven studies analyzed. Five of these studies used the DecisionDx-Melanoma test. Authors looked at the results of the seven included studies (1450 study participants) and concluded that the performance of both GEP tests (Melagenix and DecisionDx-Melanoma) varied by AJCC stage. “For patients tested with DecisionDx-Melanoma, 623 had stage I disease (six true-positive ([TP], 15 false-negative, 61 false-positive, and 541 true-negative [TN] results) and 212 had stage II disease 959 TP, 13 FN, 78 FP, and 62 TN results). Among patients with recurrence, Decision Dx-Melanoma correctly classified 29% with stage I disease and 82% with stage II disease. Among patients without recurrence, the test correctly classified 90% with stage I disease, and 44% with stage II disease.” The overall conclusion by authors was that the prognostic ability was “poor” in terms of accurately identifying recurrence in patients with stage I cancer.108
Kangas-Dick, et al. (2021) evaluated the DecisionDx-Melanoma assay in a retrospective chart review of patients at a large cancer institute. A total of 361 patients’ data were analyzed, and a follow-up period median of 15 months was established. First, sentinel node biopsy was performed for 75.9% of the patients, of which, 19.4% tested positive for recurrent cancer. Authors considered GEP class 2B status and noted that it was “significantly associated” with recurrence-free survival (RFS) and distant metastasis-free survival (MDFS) in a univariate analysis. The authors concluded that “genetic profiling of cutaneous melanoma can assist in predicting recurrence and help determine the need for close surveillance. However, traditional pathologic factors remain the strongest independent predictors of recurrence risk.”109
CP-GEP (Merlin Assay) (SkylineDx)
An eight gene expression profile test called CP [clinicopathologic]-GEP was developed to help identify patients who might safely avoid SLNB through reclassification of their risk of nodal metastasis to less than five percent. In considering certain CP variables, the developers determined that Breslow thickness and patient age were sufficiently predictive of the risk of SLN metastasis. Double-loop cross-validation [DLCV] and least absolute shrinkage and selection operator (LASSO) further identified several genes that distinguished between patients with positive and negative SLNB results. Ultimately, eight genes (MLANA, GDF15, CXCL8, LOXL4, TGFBR1, ITGB3, PLAT, and SERPINE2) along with the two CP variables (Breslow thickness and patient age) completed the CP-GEP model.
The CP-GEP model was tested in a cohort of 754 patients diagnosed with cutaneous melanoma, who underwent SLNB within 90 days of their diagnosis. The validity of the model changed based on different T categories of melanoma and demonstrated increasingly higher sensitivity with increasing T category. An inverse relationship was observed between specificity and T category. Theoretically, the model could have reduced the SNLB rate in T1b patients by 80%, though the rate reduction dropped as the T category increased across the patient population. The negative predictive value was 91% or higher across all T categories.110
The CP-GEP model was independently validated by Yousaf, et al. (2021) in a separate cohort of 208 adult patients with primary cutaneous melanoma and confirmed the trends originally published by.110 As the T category of melanomas increased, the sensitivity of the model sequentially increased, while the specificity decreased. The highest SLNB reduction rates were observed in lower T category patients (T1), and an NPV of at least 93.3% was observed across all T categories in the study. Importantly, the model was further validated focusing only on patients 65 years of age or older, as the incidence of melanoma is greater in older patient populations. Similar results were obtained compared to the full patient cohort, demonstrating the applicability of the model across all age ranges included in the study.111
11-GEP (Melagenix) (NeraCare)
Another GEP test was developed to better define the probability of patient survival at the time of first diagnosis. Eleven prognostically-relevant genes previously identified through whole-transcriptome analysis of fresh-frozen melanomas were evaluated (KRT9, DCD, PIP, SCGB1D2, SCGB2A2, COL6A6, GBP4, KLHL41, ECRG2, HES6, and MUC7), and eight of these were found to be statistically- significantly associated with melanoma-specific survival (all but ECRG2, HES6, and MUC7). Based on the coded expression data of the eight significantly associated genes, a binary GEP “score” of either zero or one was generated, representing “low risk” or “high risk”, respectively.112
To validate the GEP score, inter-assay variability was examined across four different laboratories. Concordance was high between replicates, with 93% of replicate determinations confirming the score. Next, the GEP score was challenged with a cohort of 211 melanomas whose prognostic assessment was erroneous by AJCC staging alone, where the test discriminated successfully between short-term and long-term survivors.113
An investigation by Amaral, et al. (2020) supported the clinical validity of the Melagenix test (referred to in this study as the 11-gene expression profiling score [GEPS]). GEPS was calculated for 245 patients diagnosed with stage II cutaneous melanoma, and the scores were determined to be either “high” (median: 1.06) or “low” (median: –0.21). A significant difference in melanoma-specific survival, distant metastasis free survival, and relapse-free survival was confirmed between high-score and low-score patients. Similarly, Gambichler, et al. (2021) used the 11-GEP test to determine whether a high or low score correlated with melanoma-specific survival. The authors documented decreasing probability of melanoma-specific survival with increasing 11-GEP scores. The authors further concluded that the 11-GEP is capable of identifying patients with ten year survival probabilities above 90%.115
AMBLor® test (Avero Diagnostics)
Cosgarea, et al. (2022) investigated the potential contribution of melanoma paracrine transforming growth factor (TGF)-β signaling in the loss of a protein called AMBRA1 in the skin overlying a primary tumor. The study included 109 all-stage melanoma samples, in which AMBRA1 and (TGF)-β were analyzed. Another cohort had 30 samples analyzed for (TGF)-β and 42 samples analyzed for claudin-1. Results showed “increased tumoral TGF-β2 was significantly associated with loss of peritumoral AMBRA1 (P < 0·05), ulceration (P < 0·001), AMLo high-risk status (P < 0·05) and metastasis (P < 0·01).” Although the American Joint Committee on Cancer (AJCC) staging system is an important tool, the authors noted, it leaves gaps in being unable to identify AJCC early-stage I/II tumors which are at risk of metastasis. The authors described an “urgent need” for credible prognostic information such as biomarkers in these early-stage tumors. “The combined loss of the autophagy regulatory protein autophagy-and-beclin-1 regulator one (AMBRA1) and epidermal differentiation marker loricrin (AMLo) in the tumour microenvironment as a prognostic biomarker for early-stage primary melanoma [is] associated with significantly increased risk of metastasis.”116
Labus, et al. (2020) investigated the use of the combined immunohistochemical (IHC) expression of AMBRA1 and Loricrin in the top layer of skin as a prognostic biomarker. The study was a retrospective analysis of AMBLor performed in three different cohorts of people with AJCC stage II melanomas. A clinically validated IHC assay and semi quantitative scoring method was used to define risk groups. The results showed a loss of AMBLor in the epidermis over high risk AJCC state IIA or B tumours- that was “correlated with significant reduction in disease free survival (DFS) at 12 years to 55% compared to 89% for patients with AMBLor low risk tumours (P = 0.015; HR 4.83, 95% CI: 2.29-10.14).” In addition, multivariate analysis of 80 of these tumors showed reduced disease-free survival at 12 years to 68% in the AMBLor high-risk cohort compared to 83% in AMBLor low-risk groups. The assay sensitivity was 97% and had a negative predictive value of 90%. The authors concluded that “these data indicate AMBlor as a novel prognostic biomarker for patients with non-ulcerated AJCC stage II melanomas as well as companion/stratifying biomarker for adjuvant immunotherapy.”117
Ellis, et al. (2020) sought to determine the utility of AMBRA1 and loricrin (AMLo) expression as prognostic biomarkers for AJCC stage one cutaneous melanoma. This retrospective discovery study involved 76 AJCC stage I melanoma samples. Additionally, AMLo expression was correlated against clinical outcomes for up to 12 years in two independent and retrospective validation cohorts of 379 AJCC stage I melanomas. The outcome of decreased AMBRA1 expression was a seven year disease-free survival rate (DFS) of 81.5% vs. 100% when AMBRA1 expression was maintained (P < 0·081). A combined cohort analysis also revealed a DFS rate of 98.3% in the AMLo low-risk cohort vs. 85.4% in the AMLo high-risk group. The authors concluded that “loss of AMLo expression in the epidermis overlying primary AJCC stage I melanomas identifies high‐risk tumour subsets independently of Breslow depth,” and advocated for these biomarkers to be integrated into risk stratification: “The integration of peritumoral epidermal AMBRA1/loricrin biomarker expression into melanoma care guidelines will facilitate more accurate, personalized risk stratification for patients with AJCC stage I melanomas.”118
The National Comprehensive Cancer Network
The NCCN provides common follow-up recommendations for all patients:
- “Clinical and family history can identify patients in whom multigene testing might indicate an increased genetic risk for cutaneous and uveal melanoma, astrocytoma, mesothelioma, and cancers of the breast, pancreas, and kidney. This information can guide recommendations for surveillance and early detection in appropriate patients and their relatives.
- Consider genetic counseling referral for p16/CDKN2A mutation testing in the presence of three or more invasive cutaneous melanomas, or a mix of invasive melanoma, pancreatic cancer, and/or astrocytoma diagnoses in an individual or family.
- Multigene panel testing that includes CDKN2A is recommended for patients with invasive cutaneous melanoma who have a first-degree relative diagnosed with pancreatic cancer . . .
- Testing for other genes that can harbor melanoma-predisposing mutations [see Genetic Predisposition below] may be warranted.”74
The NCCN includes the following as a risk factor for development of single or multiple primary melanomas:
- Genetic predisposition
- Presence of germline mutations or polymorphisms predisposing to melanoma (eg, CDKN2a, CDK4, MC1R, BAP1 [especially for uveal melanoma], TERT, MITF, PTEN, and potential other genes).
- Family history of cutaneous melanoma (especially if multiple); pancreatic, renal, and/or breast cancer; astrocytoma; uveal melanoma; and/or mesothelioma.”74
Regarding indications for genetic testing using emerging molecular technologies for diagnosis and prognostication, the NCCN recommended the following:
- “The panel does not recommend BRAF or NGS [next generation sequencing] testing for resected stage I-II cutaneous melanoma unless it will inform clinical trial participation.
- The BRAF mutation testing is recommended for patients with stage III at high risk for recurrence for whom BRAF-directed therapy may be an option.
- For initial presentation with stage IV disease or clinical recurrence, obtain tissue to ascertain alterations in BRAF, and in the appropriate clinical setting, KIT from either biopsy of the metastasis (preferred) or archival material if the patient is being considered for targeted therapy. Broader genomic profiling (e.g., larger NGS panels, BRAF non-V600 mutation) is recommended if feasible, especially if the test results might guide future treatment decisions or eligibility for participation in a clinical trial.
- If BRAF single-gene testing was the initial test performed, and is negative, clinicians should strongly consider larger NGS panels to identify other potential genetic targets (e.g., KIT, BRAF non-V600).”74
The NCCN also recognizes NRAS as a relevant mutation for melanoma, noting that NRAS mutations are present in “approximately 15% of melanomas with chronic and intermittent sun exposure, acral surfaces, and mucosal surfaces.” Due to the low probability of overlapping targetable mutations (such as BRAF or KIT), the NCCN remarks that “the presence of an NRAS mutation may identify patients who will not benefit from additional molecular testing.”74
NCCN acknowledges that GEP tests may have clinical utility for diagnostic purposes, noting that “melanocytic neoplasms of uncertain biologic potential present a unique challenge to pathologists and treating clinicians. Ancillary tests to differentiate benign from malignant melanocytic neoplasms include immunohistochemistry (IHC) and molecular testing via comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), gene expression profiling (GEP), single-nucleotide polymorphism (SNP) array, and next-generation sequencing (NGS). These tests may facilitate a more definitive diagnosis and guide therapy in cases that are diagnostically uncertain or controversial by histopathology. Ancillary tests should be used as adjuncts to clinical and expert dermatopathologic examination and therefore be interpreted within the context of these findings.” Similarly, NCCN comments that “noninvasive patch testing” may also be helpful to guide biopsy decisions for melanocytic neoplasms that are clinically/dermoscopically suspicious for melanoma.74
For prognostic indications, however, NCCN guidelines are more cautionary:
- “Current GEP platforms do not provide clinically actionable prognostic information when combined or compared with known clinicopathologic (CP) factors (e.g., sex, age, primary tumor location, thickness, ulceration, mitotic rate, lymphovascular invasion, microsatellites, and/or SLNB status). Furthermore, the clinical utility of these tests to inform treatment recommendations and improve health outcomes by prompting an intervention has not been established.
- Various studies of prognostic GEP tests suggest their role as an independent predictor of worse outcome. However, GEP studies to date have not demonstrated added benefit beyond comprehensive CP variables, and it remains unclear whether available GEP tests are reliably predictive of outcome prospectively collected, independent cohorts (similar to those performed in breast cancer) are necessary to define the clinical utility of molecular prognostic GEP testing as an adjunct to AJCC staging and other known prognostically significant CP variables or as part of the multidisciplinary decision-making process to guide surveillance imaging, SLBN, and adjuvant therapy.”74
The American Academy of Dermatology (AAD)
The AAD recently published guidelines for the care and management of primary cutaneous melanoma. Regarding skin biopsies, the AAD states that while many different molecular and imaging techniques have been developed, “skin biopsy remains the first step to establish a definitive diagnosis of CM [cutaneous melanoma]”; further, the guidelines also state that “Newer noninvasive techniques (eg, reflectance confocal microscopy [RCM], as well as electrical impedance spectroscopy, gene expression analysis, optical coherence tomography, and others [see the section Emerging Diagnostic Technologies]) can also be considered as these become more readily available.”119
The AAD notes that these guidelines highlight several gaps in research including “the clinical utility and prognostic significance of various biomarkers and molecular tests; optimal clinical situations in which to pursue multigene somatic and germline mutational analysis; and the value of ancillary molecular tests in comparison with well-established clinicopathologic predictors of outcome.”119 It is then noted that “Efforts to standardize the histopathologic diagnosis and categorization of melanocytic neoplasms are under way to reduce the significant interobserver variability among pathologists. Ongoing advances in genomic medicine may make many of the aforementioned issues obsolete before the next AAD melanoma CPG is issued.”119
Regarding patients with a family history of invasive cutaneous melanoma (at least three affected members on one side of the family), “Cancer risk counseling by a qualified genetic counselor is recommended.”119
Finally, the AAD states “There is insufficient evidence to recommend routine molecular profiling assessment for baseline prognostication. Evidence is lacking that molecular classification should be used to alter patient management outside of current guidelines (eg. NCCN and AAD). The criteria for and the utility of prognostic molecular testing, including GEP, in aiding clinical decision making (eg. SLNB eligibility, surveillance intensity, and/or therapeutic choice) needs to be evaluated in the context of clinical study or trial.”119
European Society for Medical Oncology (ESMO)
The ESMO has stated that “testing for actionable mutations is mandatory in patients with resectable or unresectable stage III or stage IV [melanoma] [I, A], and is highly recommended in high-risk resected disease stage IIC but not for stage I or stage IIA-IIB. BRAF testing is mandatory [I, A]. If the tumour is BRAF wild type (WT) at the V600 locus (Class I BRAF mutant) sequencing the loci of the other known minor BRAF mutations (Class II and Class III BRAF mutant) to confirm WT status and testing for NRAS and c-kit mutations are recommended [II, C].” Additionally, “patients with metastatic melanoma should have metastasis (preferably) or the primary tumour screened for detection of BRAF V600 mutation treatment options for the first- and second-line settings include anti-PD-1 antibodies (pembrolizumab, nivolumab), PD-1 and ipilimumab for all patients, and BRAFi/MEKi combination for patients with BRAF-mutated melanoma [II, B].”120
American Society of Clinical Oncology (ASCO)
The ASCO expert panel recommends needle biopsy as the preferred diagnostic approach to clinically detected lymphadenopathy in any patient with known or suspected metastatic melanoma in regional nodes. Excisional biopsy, a more invasive approach, is not routinely required for diagnosis or characterization of melanoma. The Expert Panel recommends that BRAF mutation testing should be performed at time of diagnosis, but clinicians are not required to wait for the results of that testing if the decision has been made to initiate immunotherapy. If prior testing resulted in a false negative, the panel states that re-testing RAF status upon progression could be considered, but it would not be of value as the panel is not aware of data that supports re-testing. Regarding immunohistochemistry, the panel states “Immunohistochemistry for the BRAF V600E mutation has the most rapid turnaround time; however, less common mutations may be found with other methodologies such as BRAF gene sequencing.” Beyond BRAF testing though, the Expert Panel did not make formal recommendations about other genetic testing (e.g., NRAS and KIT), given insufficient data regarding the efficacy of inhibitors targeting these pathways.121
The ASCO notes that some cancer treatments most commonly used in metastatic melanoma patients (e.g., BRAF inhibitors) may increase the risk of secondary skin cancers; hence, clinicians should be aware of this potential adverse effect when considering such therapeutic agents.122
European Dermatology Forum (EDF), the European Association of DermatoOncology (EADO), and the European Organization of Research and Treatment of Cancer (EORTC)
A panel of experts from EDF, EADO, and EORTC recommend that BRAF mutation testing be required “in patients with distant metastasis or non-resectable regional metastasis to identify those who are eligible to receive treatment with combined BRAF and MEK inhibitors, and in resected high-risk stage III melanoma patients in the adjuvant setting.”123 Mutational analysis should be performed on metastatic tissue, either distant or regional, or on the primary tumor if metastatic tissue is not feasible. Though there may be a discrepancy rate in the BRAF status between the primary versus metastatic melanoma lesions, the authors note that there is “a high concordance rate in the BRAF status…between primary and metastatic melanoma lesions.”124 Regarding next generation sequencing, the panel claims that it may help in identifying genetic alterations and, when limited to currently actionable genes like BRAF, NRAS, and KIT in a single experiment, may be cost and time effective in the clinical setting.124
The guideline also commented on NRAS, stating that NRAS mutations are present in 15-20% of melanoma cases and are almost always mutually exclusive with BRAF mutations. The guideline remarked that NRAS status may inform clinicians regarding the BRAF status, although NRAS-based treatments are still under investigation.
Regarding c-KIT, the guideline states that acral and mucosal melanomas should initially be tested for BRAF and NRAS mutations; if both genes were found to be wild-type, the sample should be tested for c-KIT.123
Tumor mutational burden (TMB) is highlighted in the guideline as a predictive tool for response to immune checkpoint inhibitors in melanoma. The panel recognized a study that demonstrated the positive association of a high TMB value with response and overall survival of melanoma patients treated with a combination of ipilimumab and nivolumab.125
Finally, the guideline comments on GEP tests like DecisionDx-Melanoma™ and their increased use by clinicians. While a growing pool of literature describes the potential application of GEP tests in the management of various cancers, the authors state “additional data are required in melanoma to address if they provide independent prognostic information in addition to known clinicopathologic factors before they can be integrated into clinical decision-making.”124
Melanoma Prevention Working Group (MPWG)
This Working Group published a guideline regarding prognostic gene expression profile [GEP] testing for cutaneous melanoma. Although the Group is “optimistic” about the future use of prognostic GEP testing to “improve risk stratification and enhance clinical decision-making”, it states that “more evidence is needed to support GEP testing to inform recommendations regarding SLNB [sentinel lymph node biopsy], intensity of follow-up or imaging surveillance, and postoperative adjuvant therapy.” Overall, the Group remarks that “there are insufficient data to support routine use of currently available prognostic GEP tests to inform management of patients with CM [cutaneous melanoma].”126
Skin Cancer Prevention Working Group (SCPWG)
An expert consensus on the appropriate use of prognostic gene expression profiling tests for the management of cutaneous melanoma was published in 2022. The SCPWG reviewed literature for DecisionDx-Melanoma, the 11-GEP (Melagenix) test by NeraCare, and the 8-GEP (CP-GEP; Merlin Assay) by SkylineDx. The eight-person consensus panel of dermatologists unanimously agreed that:
- “Gene expression profile (GEP) tests are validated, reproducible, and consistent across studies;
- Integrating GEPs into AJCC8 and NCCN models can improve prognostic accuracy;
- Prognostic GEP tests can inform clinical decision-making regarding sentinel lymph node biopsies;
- Incorporating GEPs into real-world clinical management has positively impacted patient outcomes”
Additionally, seven out of eight panelists agreed that “the AJCC8 and NCCN prognostic model, which does not account for genomic expression, may not optimize melanoma prognostic assessment.”127
The National Society for Cutaneous Medicine (NSCM)
An expert panel gathered to develop consensus-based guidelines on the appropriate use criteria for the integration of diagnostic and prognostic GEP assays into the management of cutaneous malignant melanoma. These guidelines include recommendations for the 2-GEP test (DermTech PLA), the 23-GEP test (myPath®), and the 31-GEP test (DecisionDx-Melanoma). Regarding use of the 31-GEP test, a total of 14 recommendations were considered:
A-strength recommendation:
- “Use of the 31-GEP test to aid in the management of patients who are SLNBx negative”
B-strength recommendation:
- “Integration of 31-GEP results into the decision to adjust follow-up frequency
- Integration of 31-GEP results into the decision to order adjunctive imaging studies
- Integration of 31-GEP results into management of patients with T1a tumors with Breslow depth <0.8 mm and other adverse prognostic factors
- Integration of 31-GEP results into management of patients with T1 or T2 tumors who are sentinel lymph node biopsy (SLNBx) eligible
- Integration of 31-GEP results into management of patients with T1b tumors
- Integration of 31-GEP results into management of patients with T2 tumors
- Integration of 31-GEP results into management of patients with a low-risk category based on traditional AJCC factors”
C-strength recommendation:
- “Integration of 31-GEP results into the assessment of prognosis and management options for patients with T1a tumors with a positive deep margin
- Integration of 31-GEP results into the assessment of prognosis and management options for patients with T1b tumors with a positive deep margin
- Integration of 31-GEP results to for risk-stratification of patients in clinical trials
- Use of 31-GEP results as a criterion for eligibility for a chemotherapy regimen
- T4 disease as a contraindication for use of the 31-GEP test
- Melanoma in situ as a contraindication for use of the 31-GEP test”
Seven recommendations for use of the 2-GEP test were considered as follows:
B-strength recommendation:
- “Cases in which patients present with atypical lesions requiring additional assessment beyond inspection in order to aid in the biopsy decision”
C-strength recommendations:
- Cases in which patients refuse surgical biopsy
- Cases in which suspicious lesions present in cosmetically sensitive areas
- Cases in which patients have undergone numerous biopsies in the past and wish to avoid additional biopsy procedures
- Scenarios in which patients have a relative contraindication to surgical biopsy
- Patients with increased risk of infection (e.g., immunosuppressed patients)
- Patients with heightened risk of poor wound healing
Expert consensus was reached on eight recommendations for use of the 23-GEP test:
A-strength recommendation:
- “Differentiation of a nevus from melanoma in an adult patient when the morphologic findings are ambiguous by light microscopic parameters”
B-strength recommendations:
- “Cases with pathology suggestive or suspicious for nevoid melanoma vs. benign melanocytic nevus
- Cases with pathology suggestive or suspicious for atypical Spitz tumor vs. Spitzoid melanoma
- Instances in which pathology is suggestive of or suspicious for severely atypical compound melanocytic proliferation vs melanoma on cosmetically sensitive areas”
C-strength recommendations:
- “Instances of pathology suggestive or suspicious for melanoma arising from within a severely dysplastic nevus
- Cases of pathology suggestive or suspicious for benign vs. malignant blue nevus
- To differentiate suspicious lesions in low-risk populations
- Upon request from the referring dermatologist following an ambiguous pathology report.”105
In a separate editorial published by the NSCM discussing the management of pigmented lesions, the authors conclude that “[u]se of the 2-GEP [DermTech PLA] improves patient care and improves health outcomes by using genomic assessments to minimize avoidable surgical procedures on benign lesions, enriching the population of melanomas within biopsied lesions, and decreasing downstream costs of late-stage melanoma diagnoses. For all of these reasons, creating broad access and coverage to transformative diagnostic technologies such as the 2-GEP is in the best interest of our colleagues, the healthcare system, and most importantly, our patients.”128
References
- Swetter SM. Melanoma: Clinical features and diagnosis. Updated October 4, 2023. https://www.uptodate.com/contents/melanoma-clinical-features-and-diagnosis
- Bollard SM, Casalou C, Potter SM. Gene expression profiling in melanoma: A view from the clinic. Cancer Treat Res Commun. 2021;29:100447. doi:10.1016/j.ctarc.2021.100447
- Linares MA, Zakaria A, Nizran P. Skin Cancer. Prim Care. Dec 2015;42(4):645-59. doi:10.1016/j.pop.2015.07.006
- Lee JJ, Lian CG. Molecular Testing for Cutaneous Melanoma: An Update and Review. Arch Pathol Lab Med. Oct 24 2018;doi:10.5858/arpa.2018-0038-RA
- Holmes D. The cancer that rises with the sun. Nature. Nov 20 2014;515(7527):S110-1. doi:10.1038/515S110a
- Cockerell C, Tschen J, Billings SD, et al. The influence of a gene-expression signature on the treatment of diagnostically challenging melanocytic lesions. Per Med. Mar 2017;14(2):123-130. doi:10.2217/pme-2016-0097
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. Jul 2014;38(7):934-40. doi:10.1097/PAS.0000000000000198
- Elmore JG, Barnhill RL, Elder DE, et al. Pathologists' diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ. Jun 28 2017;357:j2813. doi:10.1136/bmj.j2813
- Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. Mar 2016;74(3):395-407; quiz 408-10. doi:10.1016/j.jaad.2015.08.038
- Ransohoff KJ, Jaju PD, Tang JY, Carbone M, Leachman S, Sarin KY. Familial skin cancer syndromes: Increased melanoma risk. J Am Acad Dermatol. Mar 2016;74(3):423-34; quiz 435-6. doi:10.1016/j.jaad.2015.09.070
- Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma: a global perspective. Arch Dermatol. Nov 2001;137(11):1493-6. doi:10.1001/archderm.137.11.1493
- Hawkes JE, Truong A, Meyer LJ. Genetic predisposition to melanoma. Semin Oncol. Oct 2016;43(5):591-597. doi:10.1053/j.seminoncol.2016.08.003
- Menin C, Bojnik E, Del Bianco P, et al. Differences in telomere length between sporadic and familial cutaneous melanoma. Br J Dermatol. Nov 2016;175(5):937-943. doi:10.1111/bjd.14652
- Rossi M, Pellegrini C, Cardelli L, Ciciarelli V, Di Nardo L, Fargnoli MC. Familial Melanoma: Diagnostic and Management Implications. Dermatol Pract Concept. Jan 2019;9(1):10-16. doi:10.5826/dpc.0901a03
- Scarano C, Veneruso I, D'Argenio V. Genetic Landscape of Familial Melanoma. Genes (Basel). Jul 23 2025;16(8)doi:10.3390/genes16080857
- Harland M, Petljak M, Robles-Espinoza CD, et al. Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer. Jan 2016;15(1):139-44. doi:10.1007/s10689-015-9841-9
- Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. Sep 1994;8(1):15-21. doi:10.1038/ng0994-15
- Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. Jun 8 1995;375(6531):506-10. doi:10.1038/375506a0
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. Mar 20 1998;92(6):725-34. doi:10.1016/S0092-8674(00)81401-4
- Marzuka-Alcala A, Gabree MJ, Tsao H. Melanoma susceptibility genes and risk assessment. Methods Mol Biol. 2014;1102:381-93. doi:10.1007/978-1-62703-727-3_20
- Tsao H, McCormick SR. Inherited susceptibility to melanoma. Updated April 23, 2024. https://www.uptodate.com/contents/inherited-susceptibility-to-melanoma
- Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. Feb 2007;44(2):99-106. doi:10.1136/jmg.2006.043802
- Lynch HT, Krush AJ. Heredity and malignant melanoma: implications for early cancer detection. Can Med Assoc J. Jul 6 1968;99(1):17-21.
- Middlebrooks CD, Stacey ML, Li Q, et al. Analysis of the CDKN2A Gene in FAMMM Syndrome Families Reveals Early Age of Onset for Additional Syndromic Cancers. Cancer Res. Jun 1 2019;79(11):2992-3000. doi:10.1158/0008-5472.Can-18-1580
- Randerson-Moor JA, Harland M, Williams S, et al. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet. Jan 1 2001;10(1):55-62.
- Bishop DT, Demenais F, Goldstein AM, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. Jun 19 2002;94(12):894-903. doi:10.1093/jnci/94.12.894
- Cust AE, Harland M, Makalic E, et al. Melanoma risk for CDKN2A mutation carriers who are relatives of population-based case carriers in Australia and the UK. J Med Genet. Apr 2011;48(4):266-72. doi:10.1136/jmg.2010.086538
- Nelson AA, Tsao H. Melanoma and genetics. Clin Dermatol. Jan-Feb 2009;27(1):46-52. doi:10.1016/j.clindermatol.2008.09.005
- Wendt J, Mueller C, Rauscher S, Fae I, Fischer G, Okamoto I. Contributions by MC1R Variants to Melanoma Risk in Males and Females. JAMA Dermatol. Jul 1 2018;154(7):789-795. doi:10.1001/jamadermatol.2018.1252
- Pasquali E, Garcia-Borron JC, Fargnoli MC, et al. MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer. Feb 1 2015;136(3):618-31. doi:10.1002/ijc.29018
- Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counseling. J Am Acad Dermatol. Mar 2016;74(3):411-20; quiz 421-2. doi:10.1016/j.jaad.2015.08.037
- Simone, Valiante M, Silipo V. Familial melanoma and multiple primary melanoma. G Ital Dermatol Venereol. Jun 2017;152(3):262-265. doi:10.23736/s0392-0488.17.05554-7
- Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. Aug 28 2011;43(10):1018-21. doi:10.1038/ng.910
- Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. Feb 22 2013;339(6122):959-61. doi:10.1126/science.1230062
- Burke LS, Hyland PL, Pfeiffer RM, et al. Telomere length and the risk of cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. PLoS One. 2013;8(8):e71121. doi:10.1371/journal.pone.0071121
- Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. Oct 19 2011;480(7375):94-8. doi:10.1038/nature10539
- Yokoyama S, Woods SL, Boyle GM, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. Nov 13 2011;480(7375):99-103. doi:10.1038/nature10630
- Paszkowska-Szczur K, Scott RJ, Serrano-Fernandez P, et al. Xeroderma pigmentosum genes and melanoma risk. Int J Cancer. Sep 1 2013;133(5):1094-100. doi:10.1002/ijc.28123
- Bubien V, Bonnet F, Brouste V, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. Apr 2013;50(4):255-63. doi:10.1136/jmedgenet-2012-101339
- Gerami P, Yao Z, Polsky D, et al. Development and validation of a noninvasive 2-gene molecular assay for cutaneous melanoma. J Am Acad Dermatol. Jan 2017;76(1):114-120.e2. doi:10.1016/j.jaad.2016.07.038
- Invitae. Invitae Melanoma Panel. https://www.invitae.com/en/physician/tests/01561/
- Fulgent. Melanoma Comprehensive Panel. https://www.fulgentgenetics.com/comprehensivecancer-melanoma
- GeneDx. Melanoma Panel. https://www.genedx.com/tests/detail/melanoma-panel-831
- Berwick M, Orlow I, Hummer AJ, et al. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev. Aug 2006;15(8):1520-5. doi:10.1158/1055-9965.EPI-06-0270
- Delaunay J, Martin L, Bressac-de Paillerets B, Duru G, Ingster O, Thomas L. Improvement of Genetic Testing for Cutaneous Melanoma in Countries With Low to Moderate Incidence: The Rule of 2 vs the Rule of 3. JAMA Dermatol. Nov 1 2017;153(11):1122-1129. doi:10.1001/jamadermatol.2017.2926
- Leachman SA, Lucero OM, Sampson JE, et al. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. Mar 2017;36(1):77-90. doi:10.1007/s10555-017-9661-5
- Pissa M, Helkkula T, Appelqvist F, et al. CDKN2A genetic testing in melanoma-prone families in Sweden in the years 2015-2020: implications for novel national recommendations. Acta Oncol. Jul 2021;60(7):888-896. doi:10.1080/0284186x.2021.1914346
- Stolarova L, Jelinkova S, Storchova R, et al. Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes. Biomedicines. Oct 9 2020;8(10)doi:10.3390/biomedicines8100404
- Aoude LG, Bonazzi VF, Brosda S, et al. Pathogenic germline variants are associated with poor survival in stage III/IV melanoma patients. Sci Rep. Oct 19 2020;10(1):17687. doi:10.1038/s41598-020-74956-3
- Leong SP, Gershenwald JE, Soong SJ, et al. Cutaneous melanoma: a model to study cancer metastasis. Journal of surgical oncology. May 01 2011;103(6):538-49. doi:10.1002/jso.21816
- Yee VS, Thompson JF, McKinnon JG, et al. Outcome in 846 cutaneous melanoma patients from a single center after a negative sentinel node biopsy. Annals of surgical oncology. Jun 2005;12(6):429-39. doi:10.1245/aso.2005.03.074
- Grossmann AH, Grossmann KF, Wallander ML. Molecular testing in malignant melanoma. Diagnostic cytopathology. Jun 2012;40(6):503-10. doi:10.1002/dc.22810
- Davies MA, Gershenwald JE. Targeted therapy for melanoma: a primer. Surg Oncol Clin N Am. Jan 2011;20(1):165-80. doi:doi.org/10.1016/j.soc.2010.09.003
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England journal of medicine. Aug 26 2010;363(9):809-19. doi:10.1056/NEJMoa1002011
- Hocker T, Tsao H. Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Human mutation. Jun 2007;28(6):578-88. doi:10.1002/humu.20481
- Alrabadi N, Gibson N, Curless K, et al. Detection of driver mutations in BRAF can aid in diagnosis and early treatment of dedifferentiated metastatic melanoma. Mod Pathol. Mar 2019;32(3):330-337. doi:10.1038/s41379-018-0161-0
- Lokhandwala PM, Tseng LH, Rodriguez E, et al. Clinical mutational profiling and categorization of BRAF mutations in melanomas using next generation sequencing. BMC Cancer. Jul 5 2019;19(1):665. doi:10.1186/s12885-019-5864-1
- Burjanivova T, Malicherova B, Grendar M, et al. Detection of BRAFV600E Mutation in Melanoma Patients by Digital PCR of Circulating DNA. Genet Test Mol Biomarkers. Apr 2019;23(4):241-245. doi:10.1089/gtmb.2018.0193
- CMGL. Molecular Genetic Testing For BRAF Mutations.
- Cree IA, Deans Z, Ligtenberg MJ, et al. Guidance for laboratories performing molecular pathology for cancer patients. J Clin Pathol. Nov 2014;67(11):923-31. doi:10.1136/jclinpath-2014-202404
- Volmar KE, Idowu MO, Souers RJ, Nakhleh RE. Molecular Testing in Anatomic Pathology and Adherence to Guidelines: A College of American Pathologists Q-Probes Study of 2230 Testing Events Reported by 26 Institutions. Arch Pathol Lab Med. Sep 2015;139(9):1115-24. doi:10.5858/arpa.2014-0513-CP
- Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. The New England journal of medicine. Sep 02 2004;351(10):998-1012. doi:10.1056/NEJMra041245
- Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. The cancer journal from Scientific American. Feb 2000;6 Suppl 1:S11-4.
- Ghate S, Ionescu-Ittu R, Burne R, et al. Patterns of treatment and BRAF testing with immune checkpoint inhibitors and targeted therapy in patients with metastatic melanoma presumed to be BRAF positive. Melanoma Res. Jun 2019;29(3):301-310. doi:10.1097/cmr.0000000000000504
- Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. Aug 19 2010;363(8):711-23. doi:10.1056/NEJMoa1003466
- Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nature reviews Clinical oncology. May 24 2011;8(7):426-33. doi:10.1038/nrclinonc.2011.69
- Woodman SE, Davies MA. Targeting KIT in melanoma: a paradigm of molecular medicine and targeted therapeutics. Biochemical pharmacology. Sep 01 2010;80(5):568-74. doi:10.1016/j.bcp.2010.04.032
- NCI. Surveillance, epidemiology, and end results program. https://seer.cancer.gov/statfacts/html/melan.html
- Rashid OM, Zager JS. Genetic Testing in the Multidisciplinary Management of Melanoma. Surg Oncol Clin N Am. Oct 2015;24(4):779-93.
- O'Brien O, Lyons T, Murphy S, Feeley L, Power D, Heffron C. BRAF V600 mutation detection in melanoma: a comparison of two laboratory testing methods. J Clin Pathol. Nov 2017;70(11):935-940. doi:10.1136/jclinpath-2017-204367
- Corean JLE, George TI, Patel JL, Li KD. Bone marrow findings in metastatic melanoma, including role of BRAF immunohistochemistry. Int J Lab Hematol. Aug 2019;41(4):550-560. doi:10.1111/ijlh.13051
- Froyen G, Broekmans A, Hillen F, et al. Validation and Application of a Custom-Designed Targeted Next-Generation Sequencing Panel for the Diagnostic Mutational Profiling of Solid Tumors. PLoS One. 2016. vol. 4.
- Colombino M, Rozzo C, Paliogiannis P, et al. Comparison of BRAF Mutation Screening Strategies in a Large Real-Life Series of Advanced Melanoma Patients. J Clin Med. Jul 30 2020;9(8)doi:10.3390/jcm9082430
- NCCN. NCCN Clinical Practice Guidelines in Oncology; Melanoma: Cutaneous. https://www.nccn.org/professionals/physician_gls/pdf/cutaneous_melanoma.pdf
- Amaria RN, Prieto PA, Tetzlaff MT, et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: a single-centre, open-label, randomised, phase 2 trial. Lancet Oncol. Feb 2018;19(2):181-193. doi:10.1016/S1470-2045(18)30015-9
- Zippel D, Markel G, Shapira-Frommer R, et al. Perioperative BRAF inhibitors in locally advanced stage III melanoma. Journal of surgical oncology. Dec 2017;116(7):856-861. doi:10.1002/jso.24744
- Chiaravalloti AJ, Jinna S, Kerr PE, Whalen J, Grant-Kels JM. A deep look into thin melanomas: What's new for the clinician and the impact on the patient. Int J Womens Dermatol. Sep 2018;4(3):119-121. doi:10.1016/j.ijwd.2018.01.003
- Farmer ER, Gonin R, Hanna MP. Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol. Jun 1996;27(6):528-31. doi:10.1016/S0046-8177(96)90157-4
- Bush JW, Hunt EL, Fraga GR. BAM! Utilizing the frequency of Benign, Atypical and Malignant diagnoses for quality improvement in the histopathologic diagnosis of melanocytic neoplasms. J Cutan Pathol. Oct 2015;42(10):712-6. doi:10.1111/cup.12566
- Torres R, Lang UE, Hejna M, et al. MicroRNA ratios distinguish melanomas from nevi. J Invest Dermatol. Aug 22 2019;doi:10.1016/j.jid.2019.06.126
- Castle Biosciences. myPath® Melanoma. https://castlebiosciences.com/tests/diagnostic/mypath-melanoma/overview
- DermTech. DermTech Melanoma Test. https://dermtech.com/dermtech-melanoma-test/
- Castle Biosciences. DecisionDx®-Melanoma Overview. https://castlebiosciences.com/tests/prognostic/decisiondx-melanoma/overview
- SkylineDx. Melanoma. https://www.skylinedx.com/what/our-pipeline
- NICE. AMBLor for identifying low-risk non-ulcerated early-stage cutaneous melanomas. Updated May 5, 2022. https://www.nice.org.uk/advice/mib294/chapter/The-technology
- Clarke LE, Warf MB, Flake DD, 2nd, et al. Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J Cutan Pathol. Apr 2015;42(4):244-52. doi:10.1111/cup.12475
- Clarke LE, Flake DD, 2nd, Busam K, et al. An independent validation of a gene expression signature to differentiate malignant melanoma from benign melanocytic nevi. Cancer. Feb 15 2017;123(4):617-628. doi:10.1002/cncr.30385
- Ko JS, Matharoo-Ball B, Billings SD, et al. Diagnostic Distinction of Malignant Melanoma and Benign Nevi by a Gene Expression Signature and Correlation to Clinical Outcomes. Cancer Epidemiol Biomarkers Prev. Jul 2017;26(7):1107-1113. doi:10.1158/1055-9965.EPI-16-0958
- Castle Biosciences Inc. MyPath Melanoma. 2022;
- Minca EC, Al-Rohil RN, Wang M, et al. Comparison between melanoma gene expression score and fluorescence in situ hybridization for the classification of melanocytic lesions. Mod Pathol. Aug 2016;29(8):832-43. doi:10.1038/modpathol.2016.84
- Clarke LE, Mabey B, Flake Ii DD, et al. Clinical validity of a gene expression signature in diagnostically uncertain neoplasms. Per Med. Sep 2020;17(5):361-371. doi:10.2217/pme-2020-0048
- Varedi A, Gardner LJ, Kim CC, et al. Use of new molecular tests for melanoma by pigmented-lesion experts. J Am Acad Dermatol. Aug 13 2019;doi:10.1016/j.jaad.2019.08.022
- Ferris LK, Jansen B, Ho J, et al. Utility of a Noninvasive 2-Gene Molecular Assay for Cutaneous Melanoma and Effect on the Decision to Biopsy. JAMA Dermatol. Jul 1 2017;153(7):675-680. doi:10.1001/jamadermatol.2017.0473
- Ferris LK, Rigel DS, Siegel DM, et al. Impact on clinical practice of a non-invasive gene expression melanoma rule-out test: 12-month follow-up of negative test results and utility data from a large US registry study. Dermatol Online J. May 15 2019;25(5)doi:doi.org/10.5070/D3255044059
- Yao Z, Moy R, Allen T, Jansen B. An Adhesive Patch-Based Skin Biopsy Device for Molecular Diagnostics and Skin Microbiome Studies. J Drugs Dermatol. Oct 1 2017;16(10):979-986.
- Hornberger J, Siegel DM. Economic Analysis of a Noninvasive Molecular Pathologic Assay for Pigmented Skin Lesions. JAMA Dermatol. Sep 1 2018;154(9):1025-1031. doi:10.1001/jamadermatol.2018.1764
- Ferris LK, Gerami P, Skelsey MK, et al. Real-world performance and utility of a noninvasive gene expression assay to evaluate melanoma risk in pigmented lesions. Melanoma Res. Oct 2018;28(5):478-482. doi:10.1097/cmr.0000000000000478
- Ferris LK, Moy RL, Gerami P, et al. Noninvasive Analysis of High-Risk Driver Mutations and Gene Expression Profiles in Primary Cutaneous Melanoma. J Invest Dermatol. May 2019;139(5):1127-1134. doi:10.1016/j.jid.2018.10.041
- Skelsey MK, Brouha B, Rock J, et al. Non-Invasive Detection of Genomic Atypia Increases Real-World NPV and PPV of the Melanoma Diagnostic Pathway and Reduces Biopsy Burden. SKIN. 2021;5(5):512-523.
- Skudalski L, Waldman R, Kerr PE, Grant-Kels JM. Melanoma: How and when to consider clinical diagnostic technologies. J Am Acad Dermatol. Mar 2022;86(3):503-512. doi:10.1016/j.jaad.2021.06.901
- Robinson JK, Jansen B. Caring for Melanoma Survivors with Self-Detected Concerning Moles During COVID-19 Restricted Physician Access: a Cohort Study. SKIN The Journal of Cutaneous Medicine. 2020;4(3):248-251. doi:10.25251/skin.4.3.5
- Ribero S, Sportoletti Baduel E, Osella-Abate S, et al. Sentinel lymph node biopsy in cutaneous melanoma. G Ital Dermatol Venereol. Aug 2017;152(4):355-359. doi:10.23736/s0392-0488.16.05358-x
- Weiss SA, Hanniford D, Hernando E, Osman I. Revisiting determinants of prognosis in cutaneous melanoma. Cancer. Dec 1 2015;121(23):4108-23. doi:10.1002/cncr.29634
- Keller J, Schwartz TL, Lizalek JM, et al. Prospective validation of the prognostic 31-gene expression profiling test in primary cutaneous melanoma. Cancer Med. May 2019;8(5):2205-2212. doi:10.1002/cam4.2128
- Berman B, Ceilley R, COckerell C, et al. Appropriate Use Criteria for the Integration of Diagnostic and Prognostic Gene Expression Profile Assays into the Management of Cutaneous Malignant Melanoma: An Expert Panel Consensus-Based Modified Delphi Process Assessment. SKIN The Journal of Cutaneous Medicine. 2019;3(5)
- Marks E, Caruso H, Kurley S, et al. Establishing an evidence-based decision point for clinical use of the 31-gene expression profile test in cutaneous melanoma. SKIN The Journal of Cutaneous Medicine. 2019;3(4)
- Greenhaw BN, Covington KR, Kurley SJ, et al. Molecular risk prediction in cutaneous melanoma: a meta-analysis of the 31-gene expression profile prognostic test in 1,479 patients. J Am Acad Dermatol. Mar 27 2020;doi:10.1016/j.jaad.2020.03.053
- Marchetti MA, Coit DG, Dusza SW, et al. Performance of Gene Expression Profile Tests for Prognosis in Patients With Localized Cutaneous Melanoma: A Systematic Review and Meta-analysis. JAMA Dermatol. Sep 1 2020;156(9):953-962. doi:10.1001/jamadermatol.2020.1731
- Kangas-Dick AW, Greenbaum A, Gall V, et al. Evaluation of a Gene Expression Profiling Assay in Primary Cutaneous Melanoma. Annals of surgical oncology. Jan 23 2021;doi:10.1245/s10434-020-09563-7
- Bellomo D, Arias-Mejias SM, Ramana C, et al. Model Combining Tumor Molecular and Clinicopathologic Risk Factors Predicts Sentinel Lymph Node Metastasis in Primary Cutaneous Melanoma. JCO Precis Oncol. 2020;4:319-334. doi:10.1200/po.19.00206
- Yousaf A, Tjien-Fooh FJ, Rentroia-Pacheco B, et al. Validation of CP-GEP (Merlin Assay) for predicting sentinel lymph node metastasis in primary cutaneous melanoma patients: A U.S. cohort study. Int J Dermatol. Jul 2021;60(7):851-856. doi:10.1111/ijd.15594
- Brunner G, Reitz M, Heinecke A, et al. A nine-gene signature predicting clinical outcome in cutaneous melanoma. J Cancer Res Clin Oncol. Feb 2013;139(2):249-58. doi:10.1007/s00432-012-1322-z
- Brunner G, Heinecke A, Falk TM, et al. A Prognostic Gene Signature Expressed in Primary Cutaneous Melanoma: Synergism With Conventional Staging. JNCI Cancer Spectr. Jul 2018;2(3):pky032. doi:10.1093/jncics/pky032
- Amaral TMS, Hoffmann MC, Sinnberg T, et al. Clinical validation of a prognostic 11-gene expression profiling score in prospectively collected FFPE tissue of patients with AJCC v8 stage II cutaneous melanoma. Eur J Cancer. Jan 2020;125:38-45. doi:10.1016/j.ejca.2019.10.027
- Gambichler T, Tsagoudis K, Kiecker F, et al. Prognostic significance of an 11-gene RNA assay in archival tissue of cutaneous melanoma stage I-III patients. Eur J Cancer. Jan 2021;143:11-18. doi:10.1016/j.ejca.2020.10.016
- Cosgarea I, McConnell AT, Ewen T, et al. Melanoma secretion of transforming growth factor-β2 leads to loss of epidermal AMBRA1 threatening epidermal integrity and facilitating tumour ulceration*. British Journal of Dermatology. 2022;186(4):694-704. doi:10.1111/bjd.20889
- Labus M, Ellis R, Ewen T, et al. AMBLor: A prognostic and stratifying biomarker for adjuvant immunotherapy of AJCC stage II melanomas. Journal of Clinical Oncology. 2020;38(15_suppl):e22094-e22094. doi:10.1200/JCO.2020.38.15_suppl.e22094
- Ellis R, Tang D, Nasr B, et al. Epidermal autophagy and beclin 1 regulator 1 and loricrin: a paradigm shift in the prognostication and stratification of the American Joint Committee on Cancer stage I melanomas. Br J Dermatol. Jan 2020;182(1):156-165. doi:10.1111/bjd.18086
- Swetter SM, Tsao H, Bichakjian CK, et al. Guidelines of care for the management of primary cutaneous melanoma. J Am Acad Dermatol. Jan 2019;80(1):208-250. doi:10.1016/j.jaad.2018.08.055
- ESMO. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2025;
- Seth R, Messersmith H, Kaur V, et al. Systemic Therapy for Melanoma: ASCO Guideline. Journal of Clinical Oncology. 2020;0(0):JCO.20.00198. doi:10.1200/jco.20.00198
- Alberg AJ, LoConte NK, Foxhall L, et al. American Society of Clinical Oncology Policy Statement on Skin Cancer Prevention. JCO Oncol Pract. Aug 2020;16(8):490-499. doi:10.1200/JOP.19.00585
- Garbe C, Amaral T, Peris K, et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics - Update 2019. Eur J Cancer. Feb 2020;126:141-158. doi:10.1016/j.ejca.2019.11.014
- Garbe C, Amaral T, Peris K, et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur J Cancer. Jul 2022;170:236-255. doi:10.1016/j.ejca.2022.03.008
- Forschner A, Battke F, Hadaschik D, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J Immunother Cancer. Jul 12 2019;7(1):180. doi:10.1186/s40425-019-0659-0
- Grossman D, Okwundu N, Bartlett EK, et al. Prognostic Gene Expression Profiling in Cutaneous Melanoma: Identifying the Knowledge Gaps and Assessing the Clinical Benefit. JAMA Dermatology. 2020;156(9):1004-1011. doi:10.1001/jamadermatol.2020.1729
- Farberg AS, Marson JW, Glazer A, et al. Expert Consensus on the Use of Prognostic Gene Expression Profiling Tests for the Management of Cutaneous Melanoma: Consensus from the Skin Cancer Prevention Working Group. Dermatol Ther (Heidelb). Apr 2022;12(4):807-823. doi:10.1007/s13555-022-00709-x
- Cockerell C, Ceilley R, Lebwohl M, Rigel D. Using Genomics to Improve Pigmented Lesion Management & Health Outcomes. SKIN The Journal of Cutaneous Medicine. 2022;6(2)
- Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. Nov 2012;11(11):873-86. doi:10.1038/nrd3847
- NCI. Drugs Approved for Melanoma. Updated March 14, 2025. https://www.cancer.gov/about-cancer/treatment/drugs/melanoma
Coding Section
| Codes | Number | Description |
| CPT | 81210 | BRAF (B-Raf proto-oncogene, serine/threonine kinase) (e.g., colon cancer, melanoma), gene analysis, V600 variant(s). |
| 81272 | KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) (e.g., gastrointestinal stromal tumor [GIST], acute myeloid leukemia, melanoma), gene analysis, targeted sequence analysis (e.g., exons 8, 11, 13, 17, 18) |
|
| 81273 | KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) (e.g., mastocytosis), gene analysis, D816 variant(s) |
|
| 81311 | NRAS (neuroblastoma RAS viral [v-ras] oncogene homolog) (e.g., colorectal carcinoma), gene analysis, variants in exon 2 (e.g., codons 12 and 13) and exon 3 (e.g., codon 61) |
|
| 81345 | TERT (telomerase reverse transcriptase) (e.g., thyroid carcinoma, glioblastoma multiforme) gene analysis, targeted sequence analysis (e.g., promoter region) | |
| 81404 | Molecular pathology procedure, Level 5 (e.g., analysis of 2 – 5 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 6 – 10 exons, or characterization of a dynamic mutation disorder/triplet repeat by Southern blot analysis) | |
| 81479 | Unlisted molecular pathology procedure | |
| 81529 | Oncology (cutaneous melanoma), mRNA, gene expression profiling by real-time RT-PCR of 31 genes (28 content and 3 housekeeping), utilizing formalin-fixed paraffin-embedded tissue, algorithm reported as recurrence risk, including likelihood of sentinel lymph node metastasis |
|
| 0089U | Targeted genomic sequence analysis panel, solid organ neoplasm, DNA analysis, and RNA analysis when performed, 5 – 50 genes (e.g., ALK, BRAF, CDKN2A, EGFR, ERBB2, KIT, KRAS, NRAS, MET, PDGFRA, PDGFRB, PGR, PIK3CA, PTEN, RET), interrogation for sequence variants and copy number variants or rearrangements, if performed ***Code wording will be revised on 07/01/2025*** The following verbiage will be EFFECTIVE on 07/01/2025. Oncology (melanoma), gene expression profiling by RTqPCR, PRAME and LINC00518, superficial collection using adhesive patch(es) ***Effective on 07/01/2025*** |
|
| 0090U | Oncology (cutaneous melanoma), mRNA gene expression profiling by RT-PCR of 23 genes (14 content and 9 housekeeping), utilizing formalin-fixed paraffin-embedded tissue, algorithm reported as a categorical result (i.e., benign, indeterminate, malignant) Proprietary test: Lab/Manufacturer: |
|
| 0314U | Oncology (cutaneous melanoma), mrna gene expression profiling by rt-pcr of 35 genes (32 content and 3 housekeeping), utilizing formalin-fixed paraffin-embedded (ffpe) tissue, algorithm reported as a categorical result (i.e., benign, intermdiate, malignant) |
|
| 0387U | Oncology (melanoma), autophagy and beclin 1 regulator 1 (AMBRA1) and loricrin (AMLo) by immunohistochemistry, formalin-fixed paraffin-embedded (FFPE) tissue, report for risk of progression Proprietary test: AMBLor® melanoma prognostic test Lab/Manufacturer: Avero® Diagnostics |
|
| 0474U | Hereditary pan-cancer (eg, hereditary sarcomas, hereditary endocrine tumors, hereditary neuroendocrine tumors, hereditary cutaneous melanoma), genomic sequence analysis panel of 88 genes with 20 duplications/deletions using next-generation sequencing (NGS), Sanger sequencing, blood or saliva, reported as positive or negative for germline variants, each gene Proprietary test: GeneticsNow® Comprehensive Germline Panel Lab/Manufacturer: GoPath Diagnostics, Inc |
|
| 0578U | Oncology (cutaneous melanoma), RNA, gene expression profiling by realtime qPCR of 10 genes (8 content and 2 housekeeping), utilizing formalin-fixed paraffin-embedded (FFPE) tissue, algorithm reports a binary result, either low-risk or high-risk for sentinel lymph node metastasis and recurrence |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 02/09/2026 | Annual review, no change to policy intent. Updating policy for clairty and consistency, regulatory status, table of terminology, rationale, and references. |
| 09/25/2025 | Adding code 0578U effective 10/01/2025. |
| 05/14/2025 | Updated coding. Code 0089U verbiage will be revised on 07/01/2025. No other changes. |
| 01/22/2025 | Annual review, updating entire policy for clarity and consistency. Updating coverage criteria #1, #2 and #3. Adding new criteria #6. Adding new notes #1, #2, and #3. Also updating description rationale, references and coding. |
| 01/18/2024 | Annual review, no change to policy intent. Updating description, table of terminology, rationale, and references |
| 07/24/2023 | Annual review, merging content from CAM 257 into this policy. Updating title and verbiage to reflect that. Als updating description, table of terminology, rationale, references, notes and coding. Adding 81167, 81216, 81217, 81345, 81404, 81479, 314u and 387u |
| 11/14/2022 | Interim review to provide a positive coverage position for DermTech. Also updating description, rationale and references. |
| 04/14/2022 |
Interim review, updating note and language related to the note. No other changes. |
| 01/11/2022 |
Annual review, no change to policy intent. Updating rationale and references. |
| 01/12/2021 |
Annual review, reformatting for clarity. Adding coverage criteria for NRAS testing. Adding examples of specific tests in criteria 3. Also updating descrption, rationale, references, coding and policy number. |
| 12/10/2020 |
Updating Coding Section with 2021 codes |
| 01/06/2020 |
Annual review, policy statement added regarding expression profile testing. Also updating coding. |
| 03/06/2019 |
Corrected typo on external policy. No other changes. |
| 01/10/2019 |
Annual review, updating title for specificity, updating ICD coding. No change to policy intent. |
| 10/18/2018 |
Testing policy to correct formatting issues. No changes made to content intent. |
| 02/21/2018 |
Annual review, adding policy statement: Testing for BRAF V600 variants in patients with glioma to select patients for targeted treatment is considered INVESTIGATIONAL. Also updating background, description, regulatory status, rationale and references. |
| 04/26/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
| 03/08/2017 |
Updated Coding Section. |
| 10/18/2016 |
Annual review, no change to policy intent. Updating background, description, rationale, references and regulatory status. |
| 11/24/2015 |
Annual review, no change to policy intent. Updated background, description, regulatory status, guidelines, rationale and references. |
| 09/22/2015 |
Added ICD-10 coding to policy. |
| 07/22/2015 |
Annual review month changed to November to follow BCA review. No other changes made. |
| 08/06/2014 |
Annual review. Added related policy. Updated background, description, regulatory status, policy guidelines, rationale and references. No change to policy intent. |